Reactivity in Chemistry
Mechanisms of Glycolysis
GL5. Catalysis in Phase One
Reactions in biochemistry are usually catalysed by enzymes. In a catalysed reaction, an alternative pathway is available that makes it easier to get from reactants to products. That doesn't mean that there are fewer steps. In fact, normally there are more steps in a catalysed reaction than there are in an uncatalysed one. It does mean that the overall energy needed to traverse the catalysed barrier is lower than the energy needed to surpass the uncatalysed barrier. It's like taking the stairs up to the second floor rather than taking a running leap at the window: more steps, but overall it will save time.
So far, the reactions we have seen in glycolysis are just the overall reactions. By looking at the overall reactions, we get a pretty good sense about what is happening at each stage of the pathway. We even get some sense of how those reactions might happen, because we can identify familar nucleophiles and electrophiles that appear to be involved. Here, we will take a more detailed look at the catalytic pathways taken during the first phase of glycolysis.
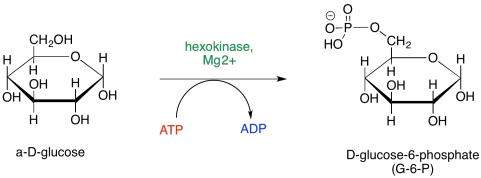
Essentially every step of glycolysis involves catalysis, and so the reactions entail cofactors and detailed steps that we have glossed over until now. The first step, phosphorylation of glucose to afford glucose-6-phosphate, requires the consumption of ATP. During that step, the terminal hydroxy group of glucose takes up phosphate from ATP, leaving ADP.
Figure GL5.1. Step 1: Glu to G6P.
It seems like that first step should be pretty straightforward, because we think of ATP as this high-energy power source for the cell, so it must be really reactive. ATP is not quite as reactive as you might think, though. That's a good thing. If it reacted too readily, it couldn't travel around the cell at all; it would get hydrolysed the first time it encountered a water molecule, and there really are an awful lot of those in a typical cell. In order to react, the ATP needs to be activated.
Part of the catalysis of the phosphorylation of glucose simply involves binding ATP to a magnesium ion. Once bound to the magnesium ion, the ATP becomes more electrophilic, because of that positive charge on the magnesium ion.
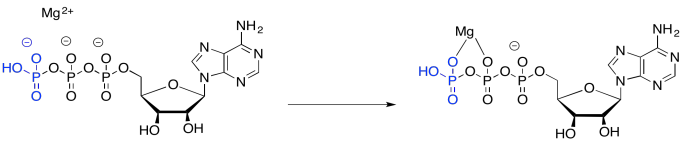
Figure GL5.2. ATP binds to Mg2+.
Although a nucleophile, such as water, is unlikely to donate to ATP -- partly because of the negative charge on the ATP -- it is more likely to donate to ATP once coordination takes place, because the magnesium ion has leveled out that negative charge.

Figure GL5.3. ATP is activated as an electrophile by binding to Mg2+.
Another aspect of catalysis in the phosphorylation of glucose involves the removal of a proton. A hydroxyl group is converted to a phosphate, and a proton is lost. Acid-base catalysis is quite common in biochemistry. There are only a handful of amino acids that commonly participate in deprotonation steps: aspartate, glutamate, lysine, and histidine. All of these residues have two structures in equilibrium: a protonated one and a non-protonated one. The non-protonated form is ready to remove a proton when needed.
Similarly, acid-base catalysis is carried out by nearby amino acid residues in the active site of the enzyme that carries out the isomerisation of glucose-6-phosphate to fructose-6-phosphate.

Figure GL5.4. Step 2: G6P to F6P.
Phosphoglucoisomerase accomplishes this task by removing a proton from an alpha position, and also from an O-H group, as well as donation of protons to a different alpha position and a different oxygen.
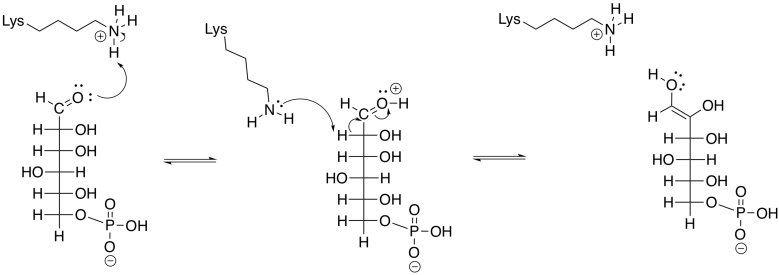
Figure GL5.5. G6P is converted to an enol.
An enol intermediate is the halfway point between the two isomers. If the proton were to be removed from that end hydroxy group, and a proton put back in the alpha position again, the structure would return to the original G6P. If, instead, a proton is removed from the second hydroxyl group along the chain, a different structure results.
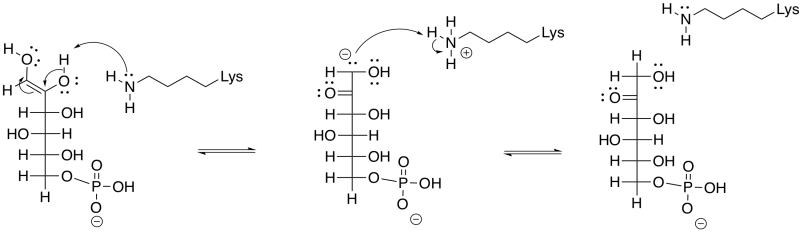
Figure GL5.6. The enol is converted to F6P.
Of course, the same sort of catalytic requirements arise again during the conversion of F6P to FBP. ATP must be activated by magnesium, and proton transfers must be carried out by acidic and basic amino acid residues.

Figure GL5.7. Step 3: F6P to FBP.
The magnesium will bind to the ATP in order to reduce the amount of negative charge. That way, the nucleophilic alcohol can donate electrons more easily. The proton from the alcohol group can be removed by an amino acid side chain, such as the negatively charged carboxylate of aspartate or glutamate, or a neutral histidine.
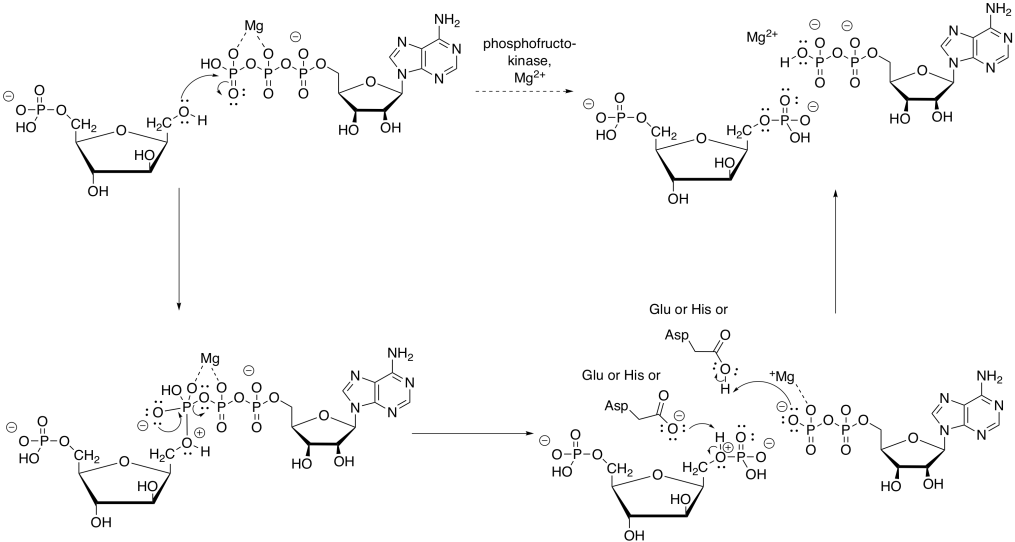
Figure GL5.8. Mechanism of the conversion of F6P to FBP.
A completely different kind of catalysis occurs during the scission step of phase one, when the six-carbon sugar is cleaved into a pair of three-carbon sugars. You may recall that this cleavage is accomplished via a retro-aldol reaction: an aldol reaction goes into reverse, spitting out an enolate or enol and a carbonyl.

Figure GL5.9. Step 4: Cleavage of FBP.
That retro-aldol step is accomplished via iminium ion catalysis. Very often in biochemical reactions, a lysine residue binds with a carbonyl to form either an iminium ion, containing an electrophilic C=N bond, or an enamine, with a nucleophilic N-C=C unit. The process begins with donation of the lysine, in its non-protonated form, to the carbonyl of FBP.
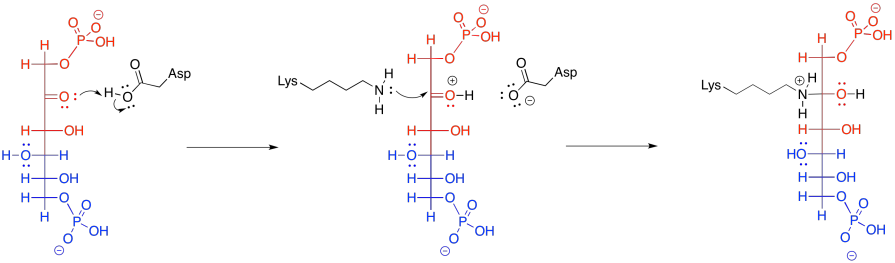
Figure GL5.10. FBP is converted to a carbinolamine.
Frequently, enzymatic reactions are presented in a more condensed form. Multiple steps are shown at once; but not just any steps. In the example below, the FBP reacts with both the lysine and the aspartate at the same time. We never draw three molecules coming together at once, because the probability of three molecules colliding at the same time is just about nil. In this case, that's not a problem; the aspartate and lysine are both part of the same molecule. Furthermore, by this point, the FBP has already bound to the enzyme, so the whole thing is one big assembly.
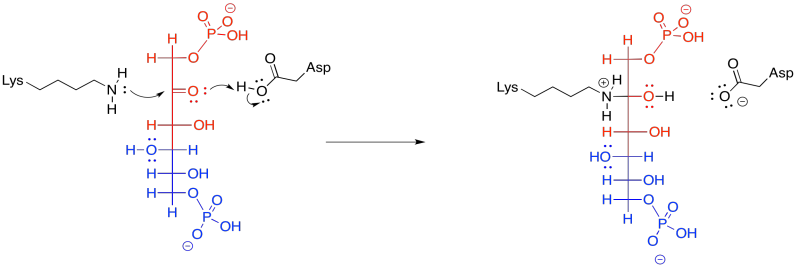
Figure GL5.11. FBP is converted to a carbinolamine in one step.
Subsequently, the carbinolamine undergoes displacement of water to yield the imine, C=N.
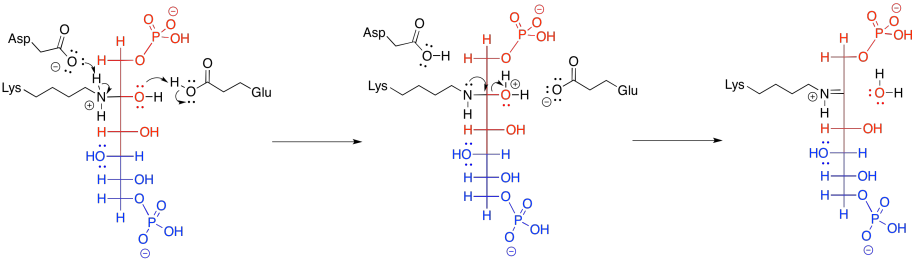
Figure GL5.12. FBP is converted to an imine.
Imines and enamines are roughly equivalent to carbonyls and enolates, respectively. Enamines are very good nucleophiles, just like enolates. Enolates are a little better, because of the negative charge. However, even though they are neutral nucleophiles, enamines are almost as good, because the reaction is driven by a less electronegative nitrogen atom; which is more willing to donate its electrons. Furthermore, in the environment of the cell, enamines form much more easily than enolates. That's because there isn't a whole lot of LDA or even NaOH floating around inside your cells. As a result, enamines are often employed as nucleophile in cases where you might think of using an enolate.
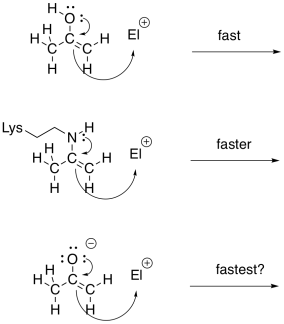
Figure GL5.13. Enols, enamines and enolates are related nucleophiles.
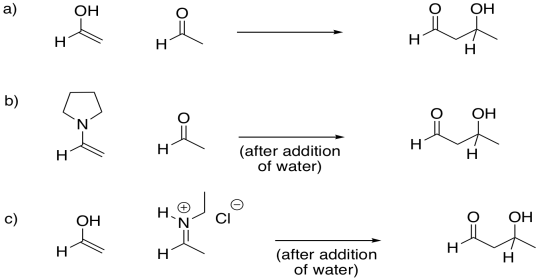
Problem GL5.1.
Provide mechanisms for the following aldol-like reactions.
The imine unit isn't an inherently better electrophile than a carbonyl; after all, it contains a less polar C=N bond instead of a C=O bond. However, the nitrogen in an imine is much more basic than the oxygen in a carbonyl. It can be protonated quite easily under biological conditions. The resulting iminium ion, containing the C=N-H+ unit, is an activated electrophile. Of course it reacts much more quickly than a regular carbonyl.
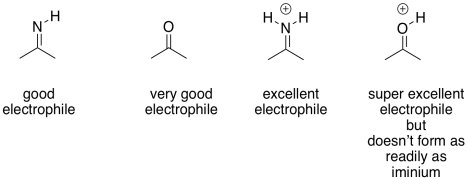
Figure GL5.14. Iminium ion is a strong electrophile.
Because carbonyls can easily form either imines or enamines, they will often be converted into those compounds in order to do reactions. This process is called enamine and imine catalysis.
Aldol reactions are actually reversible. A retro-aldol reaction is just the aldol reaction going in reverse. In that case, an enolate or an enol or an enamine might come out as a leaving group, rather than acting as the initial nucleophile.
Problem GL5.2.
Provide mechanisms for the following retro-aldol reactions.
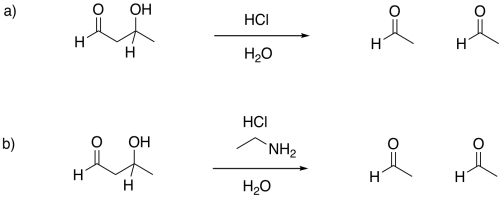
In the context of a retro-aldol reaction, we need to think about the catalysis backwards. Instead of an iminium ion acting as an activated intermediate to receive a nucleophile, it is accepting electrons to form a leaving group. Instead of having an enolate leaving group in the retro-aldol reaction, we have an enamine leaving group.
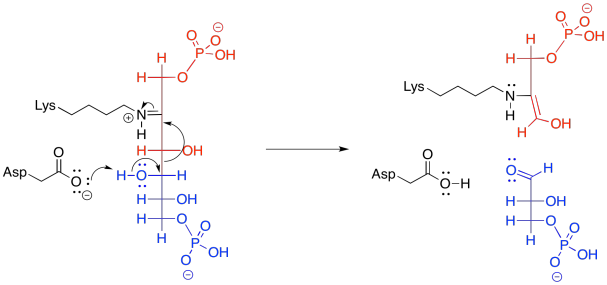
Figure GL5.15. The iminium-catalyzed retro-aldol reaction of FBP.
The lysine group, which just came on board to catalyse the reaction, is liberated by addition of a water molecule.
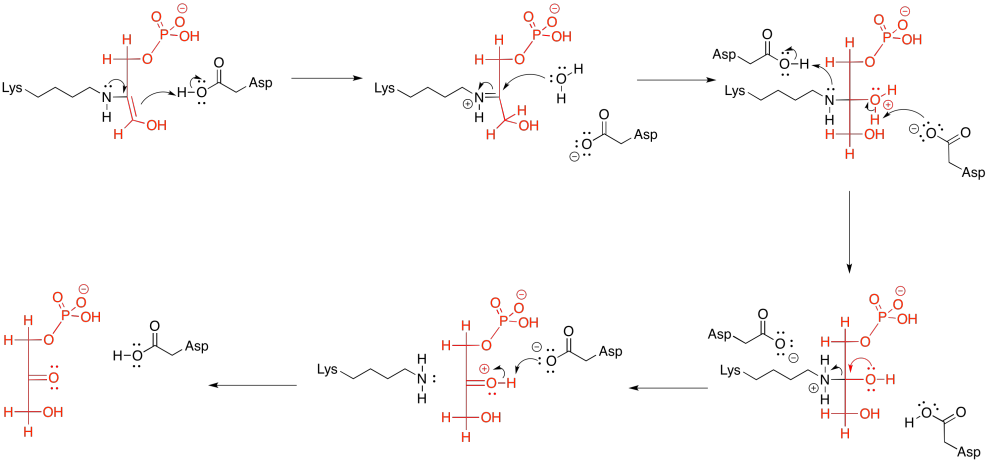
Figure GL5.16. Hydrolysis converts the imine back to a carbonyl.
There is one last reaction in phase one, the fifth overall in glycolysis. It's the conversion of DHAP to G3P; but that's just another keto-enol tautomerism. The catalytic mechanisms will be very much like those seen in the conversion of G6P to F6P.

Figure GL5.17. Step 5: DHAP is converted to a G3P.
Basic amino acid sites such as neutral histidine and anionic aspartate can readily remove a proton, whereas the corresponding conjugates can supply a proton. Tautomerism simply requires removal of a proton from an alpha position. When a proton is returned, it goes to the other end of the double bond in the enol.
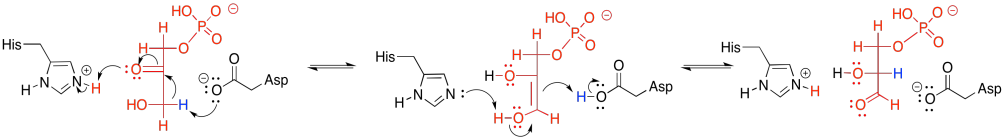
Figure GL5.18. The mechanism of conversion of DHAP to G3P.
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with other authors as noted on individual pages. It is freely available for educational use.

Structure & Reactivity in Organic,
Biological and Inorganic Chemistry by
Chris Schaller
is licensed under a
Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: