Reactivity in Chemistry
Reactions Under Orbital Control
OC8. Alkene Oxidations
There are a number of additions to alkenes that occur via concerted mechanisms. Alkene oxidations are among the most synthetically useful of these reactions because they are able to convert simple hydrocarbon starting materials into oxygen-containing compounds. The resulting heteroatomic functional groups may open up new avenues of synthetic utility or they may reflect aspects of a target natural product.
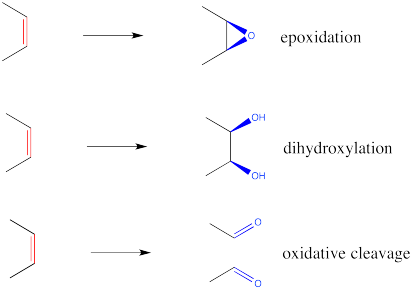
The three most common alkene oxidations are epoxidation, dihydroxylation and oxidative cleavage.
Figure OC8.1. Some common alkene oxidations.
Epoxidation
Epoxidation is a method for converting an alkene into an epoxide. The reagent required is always a peroxo species. A peroxo species looks very much like a normal oxygen-containing compound, but with an extra oxygen in it. Historically, the most common such reagent was m-chloroperbenzoic acid (mCPBA).
However, other reagents can also be used, such as hydrogen peroxide (H2O2) or potassium hydrogen persulfate (KHSO5), marketed under the trade name Oxone. The latter methods are considered "greener" or more environmentally friendly, because the side poducts (water or sulfate, respectively) are pretty innocuous. These methods are generally slower and are often used with a catalyst. Catalysts used with hydrogen peroxide include Lewis acidic species such as sodium tungstate (Na2WO4) needed to activate the peroxide. A similar reaction using titanium (IV) and chiral ligands leads to an enantiomerically pure epoxide; this reaction is called "Sharpless epoxidation". With oxone, ketones are used as oxygen transfer catalysts in a method referred to as " Shi oxidation".
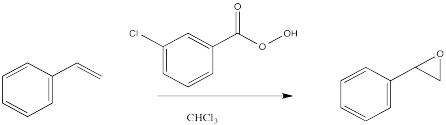
Figure OC8.2. An epoxidation using meta-chloropercenzoic acid (mCPBA).
The electrophilicity of peroxy compounds continues a theme seen in halogens such as chlorine and bromine. When two oxygen atoms are connected to each other, one of the can act as an electrophile, just as when two halogens are connected together.
During the epoxidation, the peroxy compound simply delivers its extra oxygen to the double bond. The oxygen atom both accepts a pair of electrons from the double bond and donates an electron pair to the double bond at the same time.
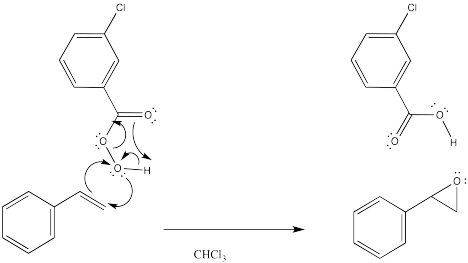
Figure OC8.3. The mechanism of epoxidation.
The reaction has something in common with pericyclic reactions. In pericyclic reactions and other reactions that take place under control of orbital symmetry, it is common to see six electrons circulating in a ring as a central feature of the mechanism. This picture is reminiscent of the aromatic structure of benzene. In fact, that aromatic stabilization is thought to play a role in stabilizing the transition states of various reactions. In this case, the three electron pairs involve delivery of the oxygen, proton transfer and π donation to the carbonyl in mCPBA. However, a similar set of arrows might not be found in the reaction of hydrogen peroxide.
Problem OC8.1.
Show how the carbonyl in mCPBA may help activate the donor oxygen toward reaction with the alkene.
Problem OC8.2.
Show how a titanium (IV) ion may help activate the donor oxygen in hydrogen peroxide toward reaction with the alkene.
The electrophilic nature of the peroxy compound is seen in the selectivity of the epoxidation reaction. Alkenes that are more electron rich tend to react much more quickly than other ones. For example, more substituted alkenes, often regarded as being electron-rich, can be selectively epoxidized in the presence of other alkenes.

Figure OC8.4.A selective epoxidation.
Furthermore, although enones can sometimes be epoxidized, the reaction is generally slower than with regular alkenes. Of course, the carbonyl attached to the alkene in an enone makes the alkene very electron-poor.
Part of the evidence for a concerted mechanism for epoxidation comes from the stereochemistry of the reaction. In general, if a cis-alkene is epoxidized, the two groups that were cis to each other in the alkene remain cis to each other in the epoxide. If the groups start out trans to each other, they remain trans in the epoxide. Just as in hydroboration, there is no opportunity for these stereochemical relationships to change.

Figure OC8.5. A stereoselective epoxidation.
Dihydroxylation
Dihydroxylation is the addition of an OH group to both sides of an alkene. Typically, when reagents such as osmium tetroxide are used, the hydroxyl groups are added to the same face of the double bond. This reaction is therefore called a syn-dihydroxylation.
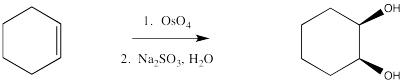
Figure OC8.6. A syn-dihydroxylation reaction.
Osmate esters can be isolated from this reaction, resulting from the concerted addition of osmium tetroxide to the alkene. Once again, this step can be compared to a pericyclic reaction. However, the osmate ester is usually decomposed in situ through the addition of a "reducing agent" such as sodium sulfite.
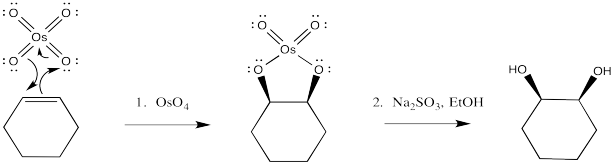
Figure OC8.7. Mechanism of syn-dihydroxylation.
Once again, the concerted nature of the reaction is seen in the stereochemistry of the product. The fact that both oxygens, which come from the osmium, are delivered to the same face of the alkene suggests that they are added at the same time.
It isn't uncommon for oxygen atoms to form additional π bonds to transition metals such as osmium. In this case, we could think of the osmium as forming an 18 electron complex as a result. Whether or not that resonance contributor is an important representation of osmium tetroxide, it is a helpful device to think of how the oxygen might form an initial attraction to the alkene.
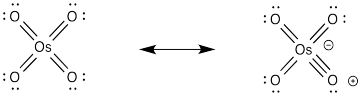
Figure OC8.8. π-Donation by oxo ligands on a transition metal.
Because of osmium tetroxide's high cost, potent toxicity and alarming propensity to rapidly sublime, other reagents are preferred. It is quite common to still use a catalytic amount of osmium tetroxide, though, along with a co-oxidant. Co-oxidants can be things like Fe(III) salts and air, although hydrogen peroxide is often used.
Oxidative Cleavage: Ozonolysis
Ozonolysis results in the complete cleavage of a double bond into two parts. The resulting fragments are each capped by an oxygen atom.
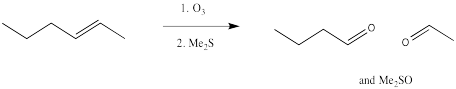
Figure OC8.9. An ozonolysis reaction.
Once again, this reaction starts out with a concerted addition of the ozone to the alkene.
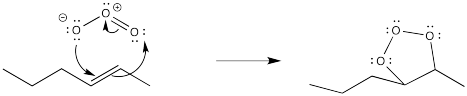
Figure OC8.10. The first step in ozonolysis involves a cycloaddition reaction.
However, the first-formed adduct, termed a molozonide, quickly rearranges to a second product, termed an ozonide. Both of these species can be isolated. However, in practice this is rarely done because of the appalling tendency of molozonides and ozonides to explode unexpectedly. The ozonide is instead decomposed through the addition of a reducing agent, such as dimethylsulfide or zinc, leaving two oxygen-containing fragments behind.
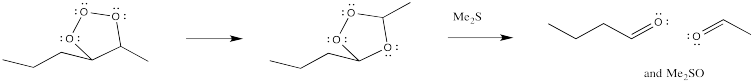
Figure OC8.11. Decomposirion of an ozonide.
Problem OC8.3.
Fill in the reagents for the following alkene oxidations.
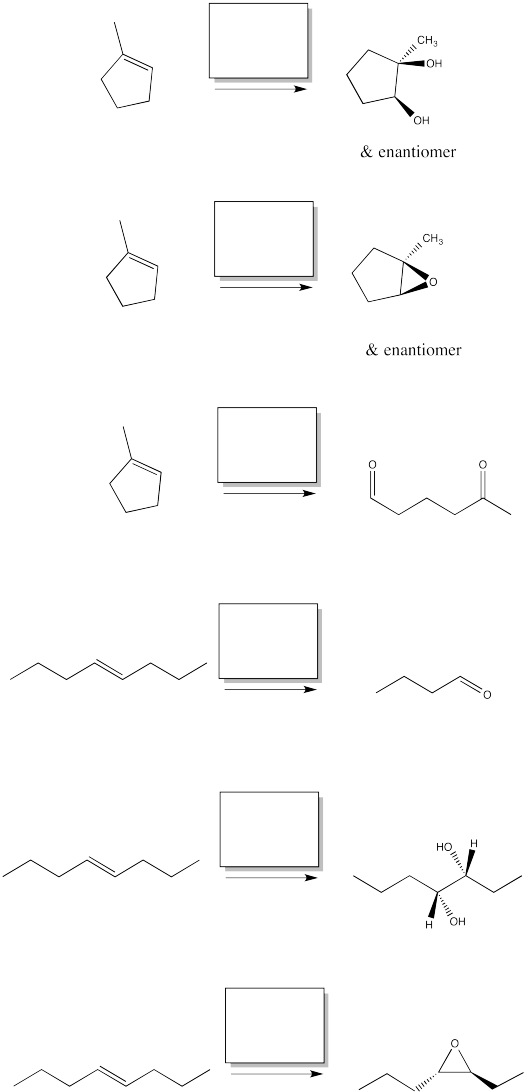
Problem OC8.4.
Fill in the products of the following alkene oxidations.
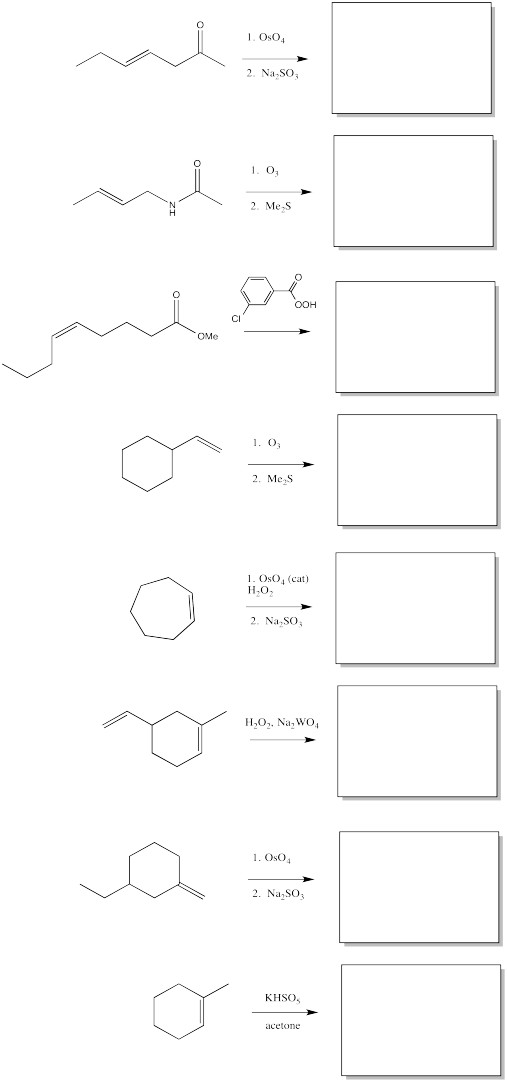
Problem OC8.5.
Fill in the blanks in the following synthesis.
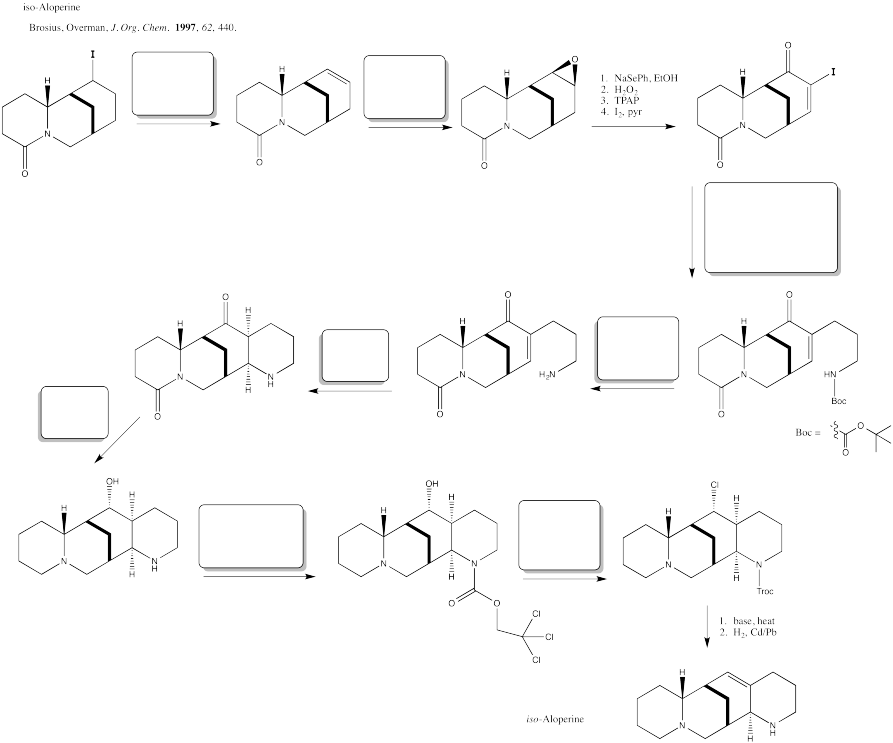
Problem OC8.6.
Fill in the blanks in the following synthesis.
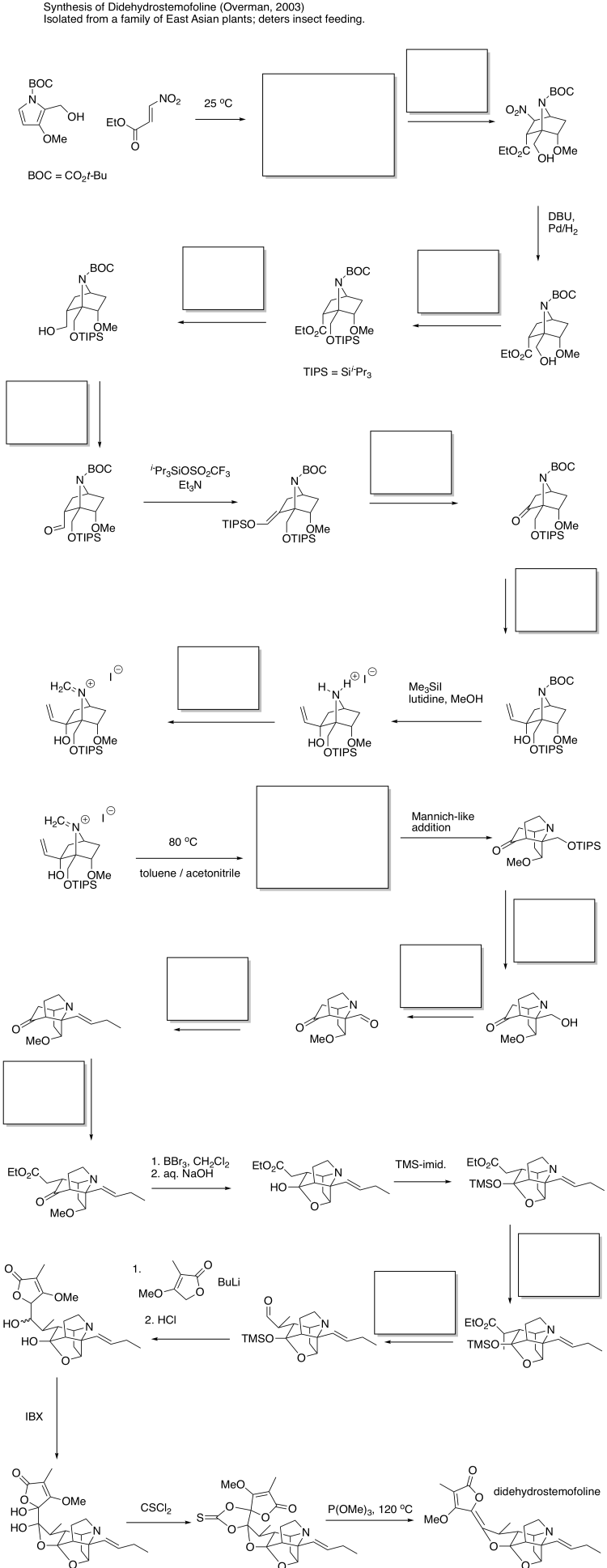
Problem OC8.7.
Fill in the blanks in the following synthesis.
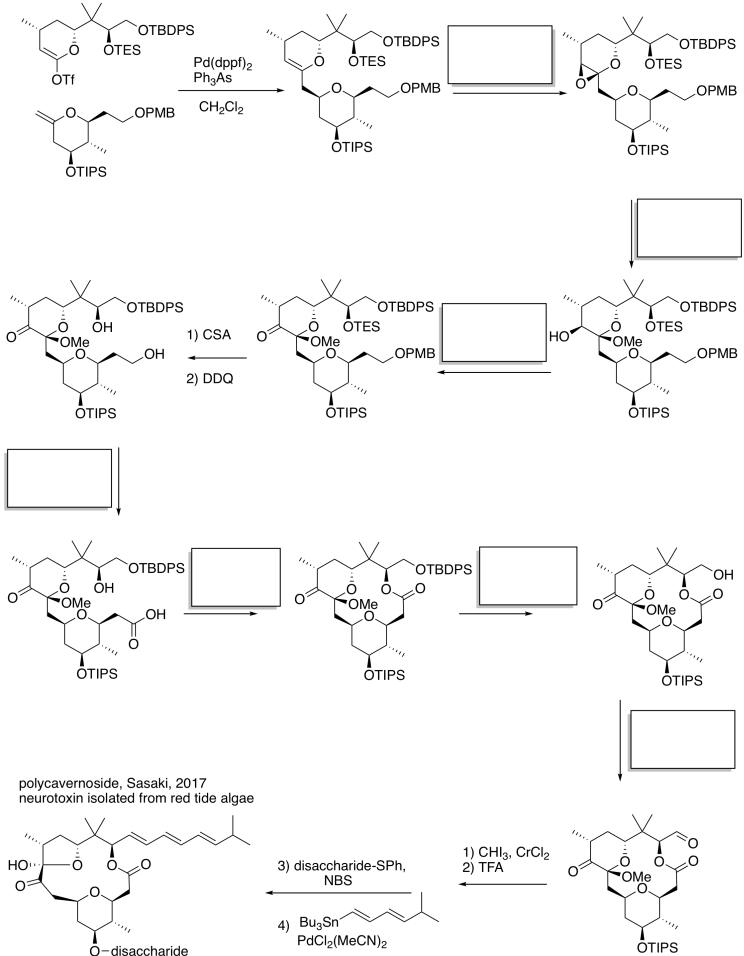
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with contributions from other authors as noted. It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: