Reactivity in Chemistry
Photochemical Reactions
PC2. Rules of Absorbance
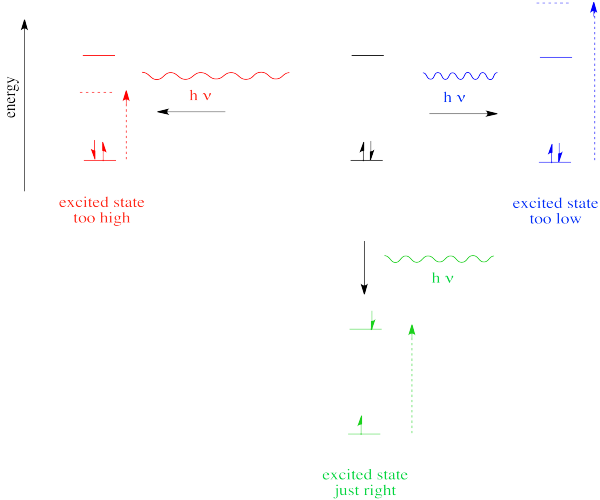
So far, we have come across one big rule of photon absorbance. In order to be absorbed, a photon's energy has to match an energy difference within the compound that is absorbing it. It can't be too big or too small; it has to be just right.
Figure PC2.1. The Goldilocks Principle of quantum mechanics.
In the case of visible or ultraviolet light, the energy of a photon is roughly in the region that would be appropriate to promote an electron to a higher energy level. Different wavelengths would be able to promote different electrons, depending on the energy difference between an occupied electronic energy level and an unoccupied one.
Other types of electromagnetic radiation would not be able to promote an electron, but they would be coupled to other events. For example, absorption of infrared light is tied to vibrational energy levels. Microwave radiation is tied to rotational energy levels in molecules.
Thus, one reason a photon may or may not be absorbed has to do with whether its energy corresponds to the available energy differences within the molecule or ion that it encounters.
Franck-Condon: Electronic and Vibrational Coupling
Photons face other limitations. One of these is a moderate variation on our main rule. It is called the Frank Condon Principle. According to this idea, when an electron is excited from its normal position, the ground state, to a higher energy level, the optimal positions of atoms in the molecule may need to shift. Because electronic motion is much faster than nuclear motion, however, any shifting of atoms needed to optimize positions as they should be in the excited state will have to wait until after the electron gets excited. In that case, when the electron lands and the atoms aren't yet in their lowest energy positions for the excited state, the molecule will find itself in an excited vibrational state as well as an excited electronic state.
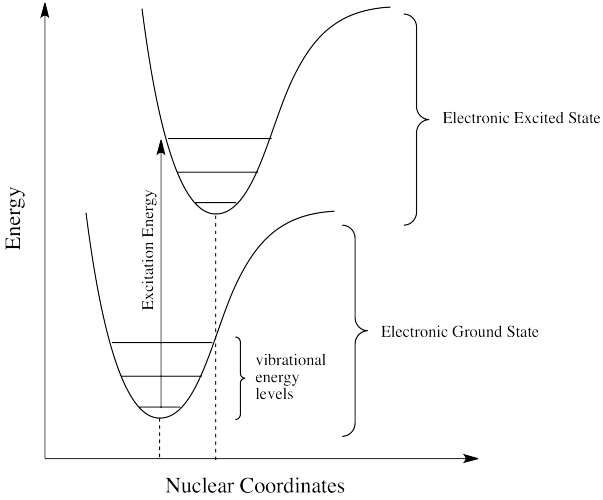
Figure PC2.2. The Franck Condon Principle.
That means the required energy for excitation doesn't just correspond to the difference in electronic energy levels; it is fine-tuned to reach a vibrational energy level, which is quantized as well.
LaPorte: Orbital Symmetry
There are other restrictions on electronic excitation. Symmetry selection rules, for instance, state that the donor orbital (from which the electron comes) and the acceptor orbital (to which the electron is promoted) must have different symmetry. The reasons for this rule are based in the mathematics of quantum mechanics.
What constitutes the same symmetry vs. different symmetry is a little more complicated than we will get into here. Briefly, let's just look at one "symmetry element" and compare how two orbitals might differ with respect to that element.
If an orbital is centrosymmetric, one can imagine each point on the orbital reflecting through the very centre of the orbital to a point on the other side. At the end of the operation, the orbital appears unchanged. That means the orbital is symmetric with respect to a centre of inversion.

Figure PC2.3. Reflection through the center of a σ orbital leaves the σ orbital unchanged..
If we do the same thing with a sigma antibonding orbital, things turn out differently.

Figure PC2.4. Reflection through the center of a σ* orbital reverses phase in the σ*> orbital.
In the drawing, the locations of the atoms are labelled A and B, but the symmetry of the orbital itself doesn't depend on that. If we imagine sending each point on this orbital through the very centre to the other side, we arrive at a picture that looks exactly the opposite of what we started with.
These two orbitals have different symmetry. A transition from one to the other is allowed by symmetry.
Problem PC2.1.
Decide whether each of the following orbitals is centrosymmetric.
a) an s orbital b) a p orbital c) a d orbital d) a π orbital e) a π* orbital
Problem PC2.2.
Decide whether each of the following transitions would be allowed by symmetry.
a) π → π* b) p → π* c) p → σ* d) d → d
Symmetry selection rules are in reality more like "strong suggestions". They depend on the symmetry of the molecule remaining strictly static, but all kinds of distortions occur through molecular vibrations. Nevertheless, these rules influence the likelihood of a given transition. The likelihood of a transition, similarly, has an influence upon the extinction coefficient, ε.
Table PC2.1. Extinction coefficients for different transitions.
| transition | ε, extinction coefficient |
| π → π* | 3,000 - 25,000 M-1 cm-1 |
| p → π* | 20 - 150 M-1 cm-1 |
| p → σ* | 100 - 7,000 |
| d → π* (MLCT) | 10,000 - 50,000 M-1 cm-1 |
| d → d | 5 - 400 M-1 cm-1 |
Problem PC2.3.
Suggest the type of transition that is probably occurring in each case.
a) A Ti(III) complex absorbs at 377 nm with ε = 95 M-1 cm-1.
b) A terpenoid ketone absorbs at 538 nm with ε = 11,500 M-1 cm-1.
c) A Ru(II) compound absorbs at 444 nm with ε = 36,000 M-1 cm-1.
Spin State
Let's take a quick look at one last rule about electronic emissions. This rule concerns the spin of the excited electron, or more correctly, the "spin state" of the excited species. The spin state describes the number of unpaired electrons in the molecule or ion.
Table PC2.2. Spin states.
| number of unpaired electrons | spin state |
| 0 | singlet |
| 1 | doublet |
| 2 | triplet |
| 3 | quartet |
The rule says that in an electronic transition, the spin state of the molecule must be preserved. That means if there are no unpaired electrons before the transition, then the excited species must also have no unpaired electrons. If there are two unpaired electrons before the transition, the excited state must also have two unpaired electrons.
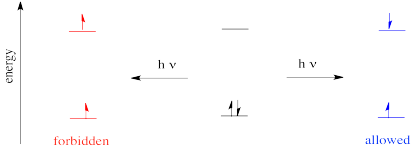
Figure PC2.5. Spin is conserved during electronic excitation.
That allowed diagram should looks strange to you. We are used to filling in electrons with spin paired for a good reason: we are following Hund's rule, that says the lowest energy state has any unpaired spins parallel, not opposite. In fact, the state on the left really is lower in energy than the state on the right; it's just that the electrons can't get from the middle state (the ground state) to the left state easily via absorption of a photon. However, they can get from the ground state to the state on the right pretty easily via photon absorption.
The state on the left is called a triplet state (the "multiplicity" of the state is the number of parallel spins plus one). The state on the right is called the singlet state. The ground state is also a singlet state. Overall, we have a ground singlet state, an excited singlet state and an excited triplet state.
As it happens, because the excited triplet state is a little lower in energy than the excited singlet state, the electron can eventually relax into the excited triplet state. It can flip, without absorbing or releasing a photon. This even is called intersystem crossing. It is related to fluorescence and phosphorescence phenomena that we will look at next.
Problem PC2.4.
Cis-trans isomerism of C=N bonds provides a path for rapid vibrational relaxation from excited states. Compounds containing this bond do not fluoresce because they are able to relax back to the ground state by transferring energy into rotational and vibrational states.
a) Illustrate this type of isomerism in the example below left.

b) Some imines (above right) are strongly fluorescent. Explain the difference.
c) Would you predict the following compound, Compound L, to absorb UV-Vis light?
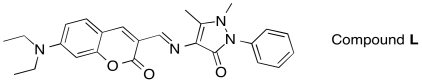
d) Would you predict Compound L to be fluorescent?
Compound L shows the absorption spectrum below (conditions: [L] = 10-6 M; cell length = 1 cm).
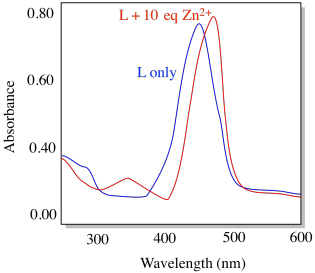
e) What is its approximate molar absorptivity at its λmax (ε, M-1 cm-1)
f) Is this transition allowed or forbidden?
g) Identify the type of transition that is likely involved.
h) In the presence of zinc ion, compound L becomes strongly fluorescent (below). Explain why with a structure. (P.-F. Wang, Org. Lett. 2007, 9, 33-36)
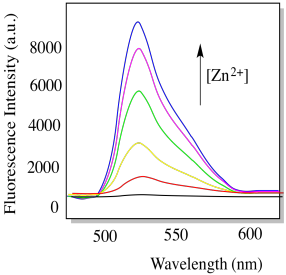
i) Propose a practical application for compound L.
Problem PC2.5.
Electron transfer between metal ions is a crucial event in a number of biochemical processes, such as oxidative phosphorylation and photosynthesis. It is thought that amino acid residues may help conduct electrons over the long distances between metals.
Harry Gray and co-workers have conducted studies of electron transfer in blue copper (azurin) proteins (J. Am. Chem. Soc. 2013, 135, 15515-15525). In the study, the protein is first tagged with [(dmp)Re(CO)3]+, which binds to a histidine residue.
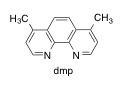
a) Draw the Re complex bound to histidine. It forms the fac isomer. Make sure you draw the ligand structures.
b) This complex is orange. What colour does it absorb?
c) If you wanted to use a laser to get the complex into an excited state, what wavelength would you choose?
d) When pulsed with a laser, the compound undergoes metal-to-ligand charge transfer (MLCT).
i) Draw the d orbital splitting diagram for this complex in the ground state.
ii) Add to your diagram a ligand orbital for MLCT at an appropriate energy level. Label it (σ, π, n etc).
iii) Show the corresponding orbital diagram for the excited state.
iv) The complex now contains a �hole� (where an electron used to be). Circle it.
e) The complex undergoes intersystem crossing to the triplet state within a picosecond (10-12 s). Show this change.
Electron transfer ensues involving the Cu(I)/(II) site (E0 = 0.3 V) and the Re complex (E0 = 1.4 V). This reaction occurs within several nanoseconds (10-9 s).
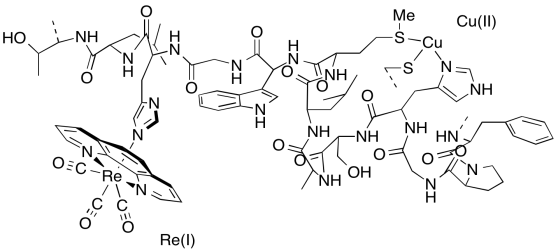
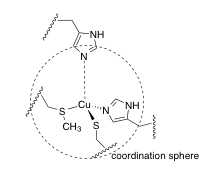
f) A portion of the X-ray crystal structure of the Cu(II) form of the protein is shown above; the structure is assumed to be similar when Cu(I) is present. Draw an arrow showing the direction of electron transfer on the structure.
g) This ET reaction is conveniently monitored by IR spectroscopy, observing the carbonyl ligands on the Re complex.
i) In what region of the IR spectrum would you observe these ligands?
ii) When electron transfer occurs, would the IR frequency shift to higher cm-1 or to lower cm-1? Explain why.
iii) The Cu-Re distance is approximately 20 Angstroms. Does electron transfer probably occur via an outer sphere mechanism or an inner sphere one?
h) In site-directed mutagenesis studies, electron transfer did not occur in a W122F mutant; i.e. with W122 (E0 = 1.37 V) replaced by F (E0 = 1.6 V). Draw a mechanism for electron transfer in the native, tagged protein that incorporates this information.
i) The reduction potential of the blue copper (II / I) site varies among different forms of the protein, from about 200 mV to 1000 mV. It has been suggested that this value is influenced mostly by amino acid residues beyond the coordination sphere. Draw in two amino acid residues that would decrease the reduction potential, and briefly state how they would do so.
Afterword: In the Re experiment, the electron eventually goes back to where it came from, but not until several microseconds (10-6 s) have passed.
This site is written and maintained by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (with contributions from other authors as noted). It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry/span> by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: