
Reactivity in Chemistry
Mechanisms of the Tricarboxylic Acid Cycle
TC3. Catalysis in the TCA Cycle
The formation of acetyl coenzyme A (acetyl CoA or AcSCoA) is step zero of the TCA cycle. It is the connecting point between glycolysis and the TCA cycle.
Figure TC3.1. Conversion of pyruvate to acetyl coenzyme A.
The sulfur in coenzyme A (CoASH) is a good nucleophile and the ketone in pyruvate is a good electrophile. Those factors should help these pieces to come together. The difficulty of this step, as we saw in the last section, is that we have no good leaving group.
Nature's approach here is to do something we call an "umpolung" approach. Umpolung means reversing polarity. We will take that good electrophile and turn it into a good leaving group. We will make it into a stable anion. In pyruvate dehydrogenase, the enzyme that catalyses this step, the umpolung effect is achieved through the use of a thiamine pyrophosphate (TPP) ylide. An ylide ("ill-id") is just a compound that contains both a positive and negative charge on atoms that are right next to each other. It has a built-in nucleophilic part and electrophilic part, and they are in close proximity to each other.
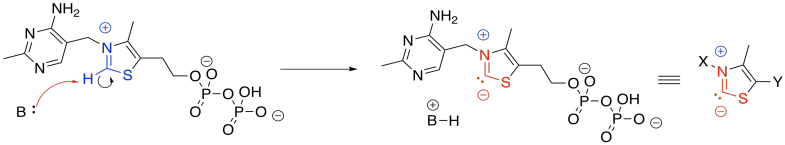
Figure TC3.2. Thiamine pyrophosphate (TPP).
The anionic position in the TPP ylide is nucleophilic. It can esily donate to that electrophilic position in pyruvate. You can see that happening in the left part of the picture above. After donation has taken place, on the right-hand side of the picture, that electrophilic carbon is no longer electrophilic. In fact, it is now found next to a C=N group. It is in an alpha position of an enamine. It is now a position of stable negative charge.
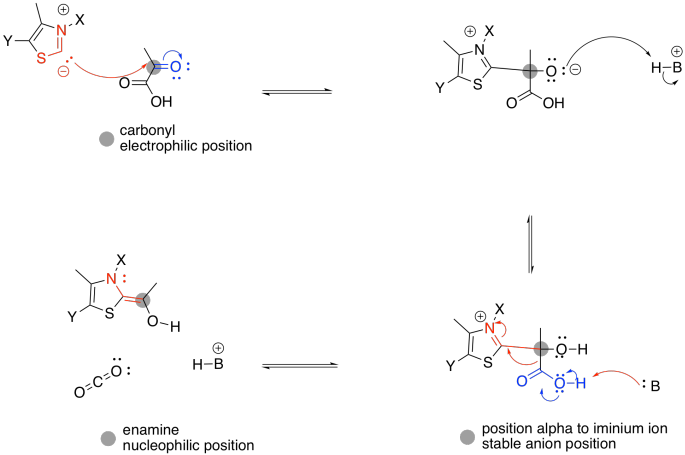
Figure TC3.3. The role of TPP in stabilizing charge.
The enamine donates to the electrophilic disulfide of lipoic acid. Don't be too surprised; sulfur can be a good nucleophile, a good leaving group, or a good electrophile, depending on its bonding situation. In this case, one sulfur atom acts as an electrophile, and the adjacent sulfur atom acts as a leaving group.
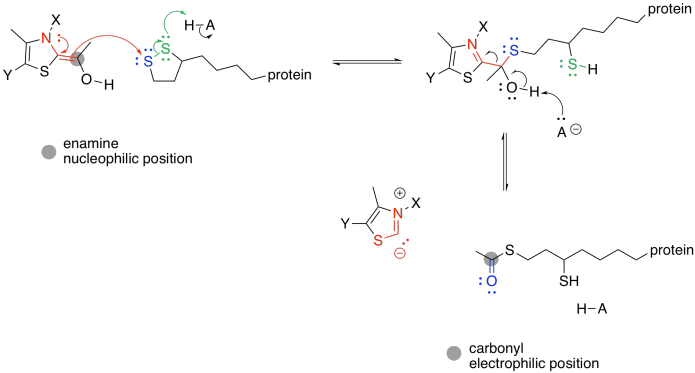
Figure TC3.4. TPP as a leaving group.
The TPP ylide is ejected via pi-donation. It is ready to be used again. At this point, the acyl group from the pyruvate has formed a thioester. Acetyl CoA is also a thioester, although it isn't the same one. An exchange of thiols accomplishes the conversion to acetyl CoA.

Figure TC3.5. Thiol exchange to form acetyl coenzyme A.
Problem TC3.1.
Provide a mechanism for the conversion of one thioester into the other. Just use generic acids and bases, H-A and B.
After formation of acetyl CoA, the new, two-carbon compound attaches to oxaloacetic acid, forming a six-carbon chain.

Figure TC3.6. Step 1: Conversion of acetyl coenzyme A to citrate.
To accomplish that step, acetyl CoA is first converted into its nucleophilic enol form. The reaction is carried out by citrate synthase. A proton is effectively transferred from the alpha position to the carbonyl oxygen. In fact, a carboxylate group in the protein removes the alpha proton and a new proton is delivered to the oxygen by another residue.
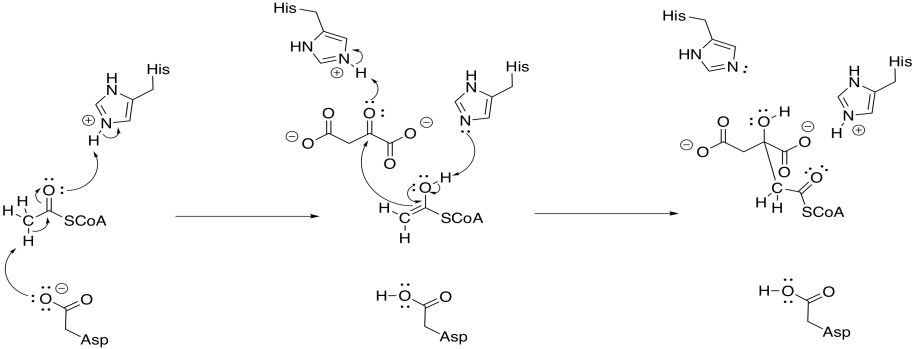
Figure TC3.7. Enolization of acetyl coenzyme A and subsequent addition to oxaloacetate.
At that point, the nucleophilic enol donates to the electrophilic ketone of the oxaloacetic acid. Picking up a proton from a nearby residue completes this straightforward addition to a carbonyl.
Problem TC3.2.
The next step is the hydrolysis of the thioester (O=C-SCoA) into a carboxylate ion (O=C-O-). Provide a mechanism for this step. Use amino acid residues for acid and base steps.
Aconitase carries out the next stage of the cycle, an isomerisation from citrate to isocitrate, in which a hydroxy group moves from one carbon to the next.

Figure TC3.8. Step 2: Conversion of citrate to isocitrate.
Aconitase is an unusual enzyme because it contains an iron-sulfur (FeS) cluster, a cubic group of four iron and four sulfur atoms, coordinated to a protein (usually through cysteine residues). Lots of enzymes contain FeS clusters, but usually they are concerned with shuttling individual electrons from one place to another. Aconitase doesn't do that, so why does it have its own FeS cluster?
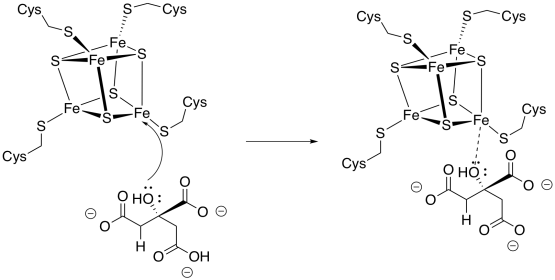
Figure TC3.9. Citrate binding to an FeS cluster.
The isomerisation step occurs with specific stereochemistry, and it is believed that the FeS cluster plays a role in holding the molecule in a specific orientation while the reaction takes place. Usually, it is assumed that the hydroxy group coordinates with one of the iron atoms, but coordination by the carboxylates is also possible.
Once the citrate is in the proper position, the original hydroxy group is removed in a dehydration step. Here, the FeS cluster is left out of the picture to simplify things. The hydroxy group is protonated by a histidine residue, making a better leaving group, while a deprotonated serine residue pulls a proton from the nearby alpha position to complete the dehydration. It's unusual to see a deprotonated serine, but it is probably employed here because it is a very strong base. Other amino acids nearby probably play a role in stabilising that serine anion through intermolecular forces so that it can form more easily.
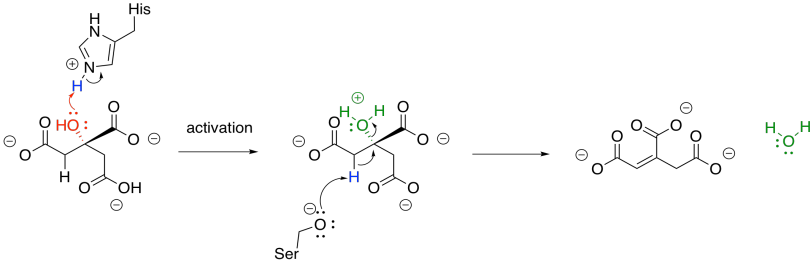
Figure TC3.10. Elimination of water from citrate.
The sequence of some of these steps is not always clear, and sometimes appear to occur so quickly as to be simultaneous. That speed is made possible by the close proximity of the reactants in the active site of the protein. The step shown above is laid out as a separate activation step and deprotonation step in order to make the role of each amino acid very clear, but they don't necessarily happen one after the other like that.
Once the first water molecule is gone, we need to add the second to the other side of the new double bond. This step occurs via a 1,4-addition, and is still catalysed by aconitase. The 1,4-addition would result in an enolate anion, which would then be protonated. In the drawing, the steps are condensed, so the nearby histidine is already protonating that alpha position before the anion forms.
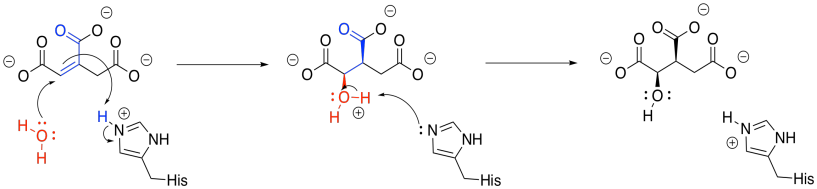
Figure TC3.11. Addition of water to form isocitrate.
The isocitrate goes on to an oxidative decarboxylation step, carried out by isocitrate dehydrogenase.

Figure TC3.12. Step 3: Conversion of isocitrate to α-ketoglutarate.
This enzyme requires a magnesium ion cofactor that binds and activates the isocitrate. There are always different ways to draw complexes like this. We can think of the isocitrate sort of sticking to the magnesium; we can think of it as forming one ionic bond and one dative bond; and we can think of that dative bond as conferring a positive charge on the hydroxyl oxygen, which has donated a lone pair. That's a useful way to think about it this time, because we are going to remove that proton from the hydroxy group.
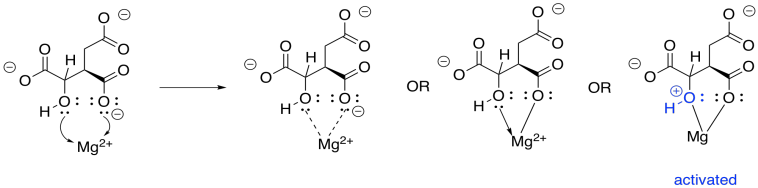
Figure TC3.13. Coordination of isocitrate to Mg2+.
In this step, we face that problem of removing two protons and two electrons from the molecule, converting an alcohol to a ketone. Stated differently, we need to lose a proton (H+) and a hydride ion (H-). We need a base to remove the proton, and in this case a "relay" system is used, in which a protein residue is sitting a little too far away to pluck off that proton, so it takes one from a water, and the water takes one from the hydroxy group.
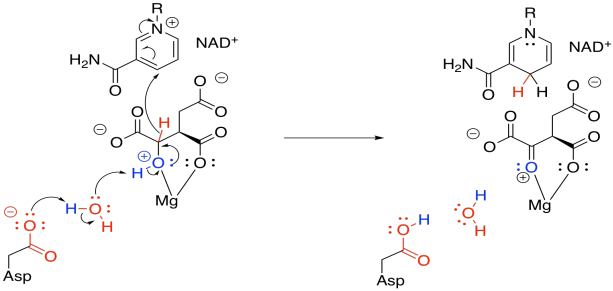
Figure TC3.14. Oxidation of coordinated isocitrate.
The hydride is delivered to NAD+, a biochemical oxidising agent. It's frequently used to accept a pair of electrons in the form of a hydride ion. The NAD+ is converted to NADH, and the NADH stores that electron pair for use in other processes.
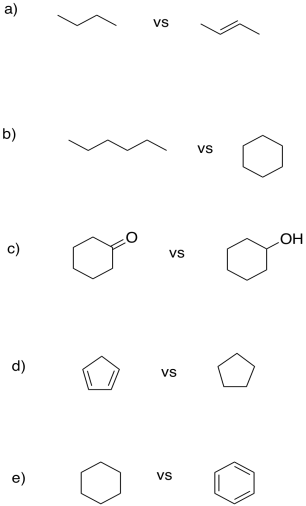
Problem TC3.3.
"Units of unsaturation" refer to how one molecule differs from another only by loss of groups of H2 (or two hydrogen atoms, or two protons and two electrons). In each case, show how many protons and electrons are needed to convert one compound into the other.
Now that the ketone has been formed, loss of carbon dioxide from the alpha position is relatively easy, because the CO2 leaves behind a stable enolate anion. In this case, the enolate is further stabilised by the magnesium ion. The enolate is neutralised by picking up a proton from a nearby tyrosine. The compound that forms is called alpha-ketoglutarate.
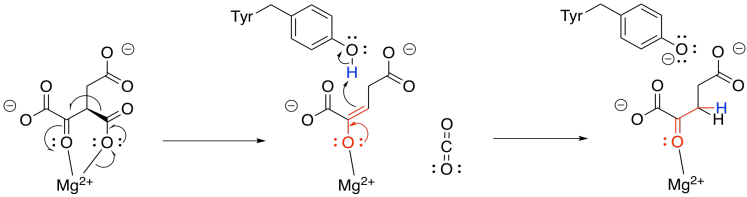
Figure TC3.15. Decarboxylation to form α-ketoglutarate.
Problem TC3.4.
Explain why a tyrosine residue can be deprotonated very easily, but it is more difficult to remove a proton from a serine residue.
The alpha-ketoglutarate undergoes a second decarboxylation, carried out by alpha-ketoglutarate dehydrogenase. In the equation below, the carbon dioxide is lost from the left-hand side of the alpha-ketogllutarate molecule.

Figure TC4.16. Step 4: Conversion of α-ketoglutarate to succinyl coenzyme A.
Decarboxylation requires a stable anion as a leaving group from the CO2. In the case of isocitrate, that stable leaving group was provided by installing a carbonyl. The alpha-ketoglutarate already has a carbonyl, but it's in the wrong place. It's right next to the carboxylate group instead of one carbon over. This situation is similar to the one in which pyruvate was decarboxylated to form acetyl CoA, and it is solved in exactly the same way, using a TPP ylide. The TPP ylide installs an iminium group one carbon away from the carboxylate, meaning there is now an ability to stablise an anion at the right place.
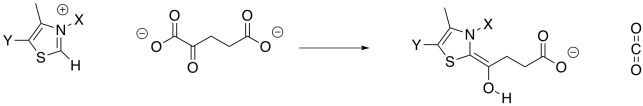
Figure TC3.17. Addition of a TPP ylide aids in decarboxylation.
Problem TC3.5.
Show the mechanism of TPP addition to alpha-ketoglutarate, and the subsequent decarboxylation. You can use generic B: and A-H as catalysts.
As in the case of pyruvate dehydrogenase, the TPP unit must be removed again. However, it is now part of a nucleophilic enamine group, and the obvious reaction partner for it is an electrophile. An electrophilic disulfide in lipoic acid is again used to reform the iminium group.
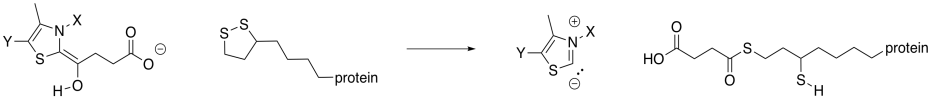
Figure TC3.18. Removal of the TPP ylide.
Problem TC3.6.
Show the mechanism of reaction with lipoic acid.
The ring-opened lipoic acid is traded for coenzyme A; that's just one sulfur nucleophile for one sulfur leaving group.
If you pay attention to the map of the citric acid cycle, you see that this step consumes another NAD+ molecule, and results in another NADH. From the point of view of the main reactant that we are following through the cycle, we have nothing left to do. The alpha-ketoglutarate has been converted to succinyl coenzyme A, which is waiting for the next step. The NAD+ is needed in a separate catalytic cycle, in which the ring-opened lipoic acid is closed again to form a new disulfide. The disulfide will be ready to react and keep the TCA cycle going.
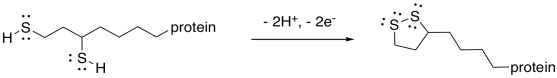
Figure TC3.19. Oxidation of thiols to form a disulfide linkage.
This mechanism is actually a little bit complicated and requires shuttling of protons and electrons from one place to another. A second biochemical oxidising agent, FAD, acts as an intermediary that transports these pieces to the NAD+.
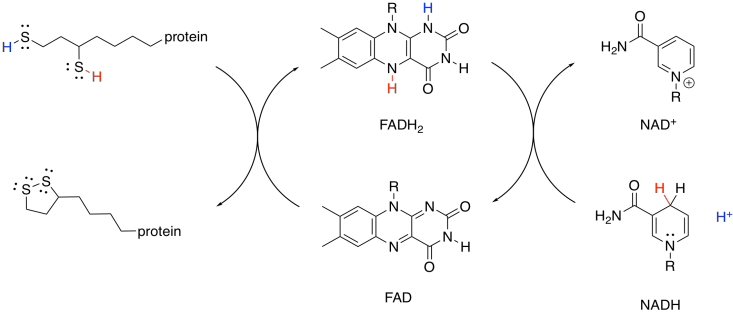
Figure TC3.20. FAD mediates the oxidation of the dithiol by NAD+.
The ATP-forming step of the citric acid cycle takes up an inorganic phosphate and couples it to guanosine diphosphate; this step is carried out by succinyl coenzyme A synthetase. The guanosine diphosphate transfers the phosphate to an ADP in a separate cycle.

Figure TC3.21. Step 5: Conversion of succinyl coenzyme A to succinate.
The trouble with phosphate-phosphate coupling is all of that negative charge, and no good leaving groups. Presumably, if we add another phosphate to a GDP or an ADP, we have displaced a hydroxide ion (HO-) or, even worse, an oxide ion (O2-). So, we need a work-around to take care of that problem.
The negatively charged phosphate can certainly act as a nucleophile. It displaces CoASH from succinyl coenzyme A, forming an activated phosphoanhydride.
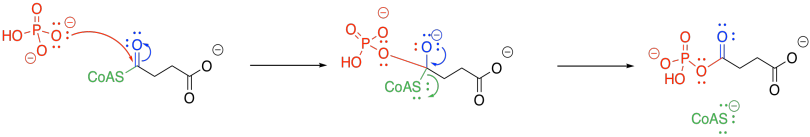
Figure TC3.22. Activation of succinyl coenzyme A as a phosphoanhydride.
The phosphate group is transferred to a histidine. The histidine is nucleophilic, so it can donate to the phosphoanhydride group. An oxygen is lost from the phosphorus, forming a phosphonate group. The succinate is displaced as a leaving group, where it waits for the next part of the TCA cycle.
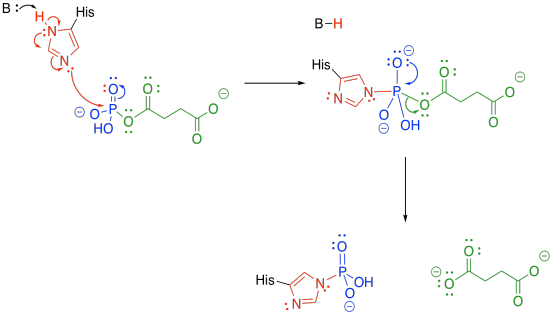
Figure TC3.23. A dephosphorylation step follows.
Meanwhile, the phosphate-histidine conjugate delivers the phosphonate group to guanosine diphosphate. The histidine is displaced as a leaving group, and is ready to start its cycle again.
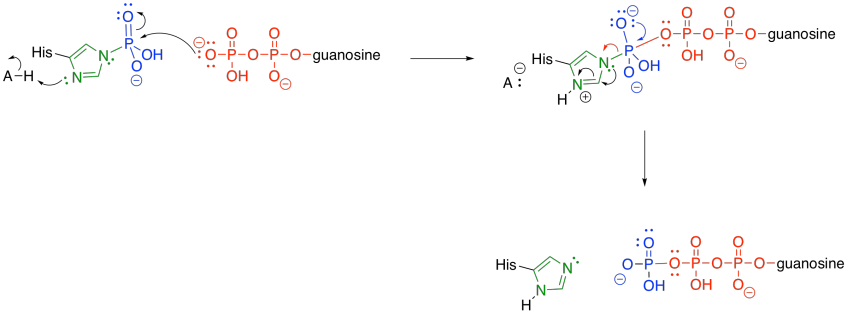
Figure TC3.24. Dephosphorylation of the catalytic histidine
At this point, just a few steps are needed to regenerate the oxaloacetate in order to start the TCA cycle over again. Some of those steps are still very important in terms of energy packaging for the cell. For example, the next step is catalysed by succinate dehydrogenase, which is an integral part of the oxidative phosphorylation complex in most species (that complex carries out the oxygen-consuming part of respiration).

Figure TC3.25. Step 6: Conversion of succinate to fumarate.
By now, you may have noticed that a dehydrogenase makes use of those biochemical oxidants, FAD and NAD+. This time, FAD is involved. Once again, two protons and two electrons need to be removed from succinate in order to form fumarate. And, once again, we can think about that as a proton and a hydride ion. A proton is removed from the alpha position, and the resulting enolate ion ejects a hydride onto the waiting FAD molecule.
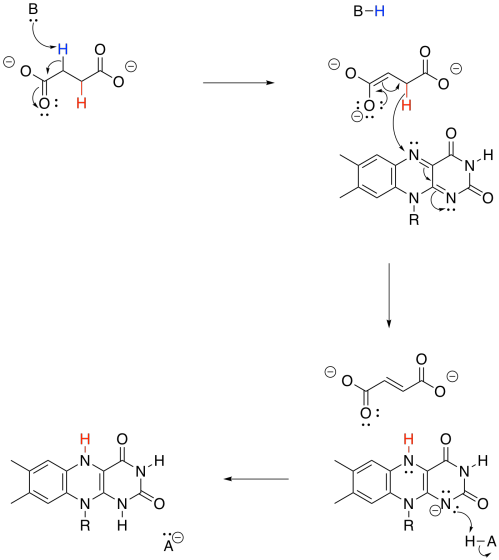
Figure TC3.26. Oxidation of succinate by FAD.
The conversion of fumarate to malate makes use of the enzyme, fumarase. The reaction is a hydration, similar to the one we saw carried out by aconitase. Note that a water molecule is added to one end of the double bond, which is electrophilic because it is conjugated with a carbonyl.

Figure TC23.27. Step 7: Conversion of fumarate to malate.
Problem TC3.7.
Provide a mechanism for conversion of fumarate to malate.
Finally, malate is turned into oxaloacetate through the action of malate dehydrogenase. A NAD+ cofactor is required for this step. Like some other steps we have seen before, this one involves the conversion of an alcohol group into a ketone group. It involves the loss of a proton and a hydride ion.

Figure TC2.28. Step 8: Conversion of malate to oxaloacetate.
Problem TC3.8.
Provide a mechanism for conversion of malate to oxaloacetate.
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with other authors as noted on individual pages. It is freely available for educational use.

Structure & Reactivity in Organic,
Biological and Inorganic Chemistry by
Chris Schaller
is licensed under a
Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: