04/23/2007
| The following is from the
WebMD
Health "In general, people with diabetes either have a total lack of insulin (type 1 diabetes) or they have too little insulin or cannot use insulin effectively (type 2 diabetes).
How are these diseases different?
How are they alike?Both types of diabetes greatly increase a person's risk for a range of serious conditions. Despite the fact that monitoring and management of the disease can usually prevent most complications, diabetes remains the leading cause of blindness and kidney failure. It also continues to be a critical risk factor for heart disease, stroke, and leg amputations." |
We will focus our attention only on Type II Diabetes, the occurrence of which is increasing so dramatically around the world that it has become pandemic.
What causes the disease? No one knows for sure but it is clearly associated with obesity. More than 3/4 of all people with Type II diabetes (henceforth referred to only as diabetes) are obese. The doesn't not imply that obesity causes diabetes (as we learned from our study of correlation vs causation), but at a minimum, it appears to be a highly contributive factor. In the US, minority ethnic and racial groups (who also tend to be poorer and have less education) have a higher percentage of people with the disease. Several factors have been cited included genetic predisposition, higher consumption of prepared "junk" food, and relative lack of health care and health insurance.
| group (US) | % diabetic (US) |
| Non-Hispanic white | 8 |
| Non-Hispanic black | 13 |
| Hispanic/Latino | 10 |
| American Indians/Alask Natives | 15 |
| reference: http://www.healthlit.org/pdfs/DiabetesBook8x.pdf | |
Diabetes is diagnosed by a fasting (8-10 hr) blood sugar measurement. Normal people have levels from 70-110 mg/dl. Greater than 126 suggests diabetes. People with Type II diabetes, in contrast to those with Type I, can make insulin. Insulin is a small protein secreted from pancreatic islet cells in response to increased blood sugar. Type II diabetics either don't secrete enough insulin, or more likely, it doesn't work as well as it should in promoting glucose uptake into cells (which of course increases blood glucose level). What is insulin and how does it work?
Insulin
Insulin is a small protein. It is synthesized as one chain, which is then cleaved at one site by a protease to form a two chain (A and B) protein. The two chains are covalently linked by a bond (disulfide) between a cysteine, an amino acid with a -CH2SH side chain, with a cysteine side chain on the other chain. This protein binds to its receptor, the insulin receptor, on cell membranes, which initiates a process of signal transduction.
We have studied membrane protein receptors before. One was the glucagon and epinephrine receptors, which when bound by the hormone led to activation of adenylate cyclase in the cell, generating the second messenger, cyclic AMP (cAMP). This then bound to and activated Protein Kinase A (PKA) which started a cascade of protein phosphorylations ending in the phosphorylation and activation of the enzyme, glycogen phosphorylase, which cleaved glucose from the storage form of glucose in liver and muscle, glycogen. We also studied neurotransmitter receptors in post-synaptic neurons. In these cases, the neurotransmitter usually gated the receptor open which caused ion flow across the membrane, changing membrane transmembrane potential. In some cases, the neurotransmitter bound to its receptor, which led to protein phosphorylation within the neuron.
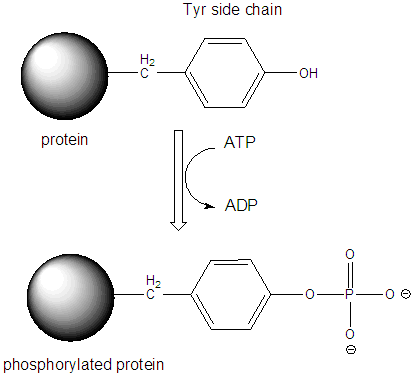
What happens when insulin binds it receptor? Its extremely complicated, but the initial event is perhaps easier that what is described above for the case of epinephrine and glucagon. In that case a second messenger (cAMP) was made which then bound to PKA and activated it. Why not skip the intermediate step in the activation? Wouldn't it be easier if the insulin receptor, when bound to insulin, becomes an active kinase on its own? That is exactly what happens: the insulin receptor is a hormone-dependent kinase. When insulin binds on the extracellular part of the insulin receptor, shape changes communicated to the intracellular part cause it to bind ATP and phosphorylate proteins, specifically on tyrosine side chains. The insulin receptor is an insulin-dependent tyrosine kinase. The first protein that the insulin receptor phosphorylates is itself. This then allows other intracellular proteins to bind to the intracellular domain of the receptor, and become phosphorylated.
![]() Chime Model:
Insulin
Chime Model:
Insulin ![]()
![]() Chime Model:
Insulin Receptor Tyrosine Kinase
Chime Model:
Insulin Receptor Tyrosine Kinase ![]()
What happens next? Obviously, the intracellular proteins that are phosphorylated by the activate insulin receptor would be prime candidates for investigation. One of the proteins is called the insulin receptor substrate 1 (IRS-1). A cascade of phosphorylations and shape/activity changes ensue. Before we study that, lets see what happens in general to cells when insulin binds. Since carbohydrate and lipid metabolism are linked, as well as diabetes and obesity, we will consider the effect of insulin on the liver (where glycogen is stored and broken down, and glucose is synthesized) and muscle (where glycogen is stored and broken down, and much glucose is used to produce energy in the form of ATP) and fat tissue (where fat is stored and broken down). The liver in times of low energy oxidizes fatty acids for energy. The glucose made new and from breaking down glycogen reserves is send into the blood for use by the brain and other peripheral organs.
Muscle and liver: Insulin promotes glucose entry into muscle cells. Glucose is a very polar molecule, which could not easily diffuse through the nonpolar lipid bilayer without help. That help comes from a membrane protein, a glucose transporter encoded by the GLUT4 gene, through a process called facilitated diffusion. The protein is sequestered in small vesicles in the cytoplasm and on insulin binding to the cell membrane, signal transduction processes cause the vesicle to fuse with the cell membrane, bringing the glucose receptor to the surface. The liver (which also stores glucose as glycogen) and brain use another facilitated transporter to bring glucose into the cell, so insulin is not needed.
animation: Glut4 movement to membrane (in this case a fat cell)
When glucose gets into muscle and liver cells, the predominate result is the synthesis of glycogen, allowing the storage of the energy in glucose until it is needed in the future. Insulin in effect signals the "well fed" state, and promotes complex carbohydrate synthesis and storage. When insulin levels go down, the effect is reversed, and glycogen breakdown ensues.
Fat Tissue: To see what happens in the fat cells, we must first consider what happens in the liver. When glucose and insulin levels are high, the liver synthesizes glycogen. Eventually the liver glycogen concentrations are maximized, and it uses excess glucose to synthesize fatty acids (remember, sugar can be converted to fatty acids but not the reverse). These get synthesized into triacylglycerides which are packaged by the liver in particles called lipoproteins which enter the blood. When they arrive at fat tissue, the fatty acids are cleaved off and enter the fat cell. Insulin, as it does in the muscles, stimulates glucose entry into fat cells. The glucose in the fat cells starts off down the glycolytic pathway but doesn't go all the way. It stops at a C3 molecules (glyceraldehyde) to which the incoming fatty acids covalently attach to form triacylglycerides.
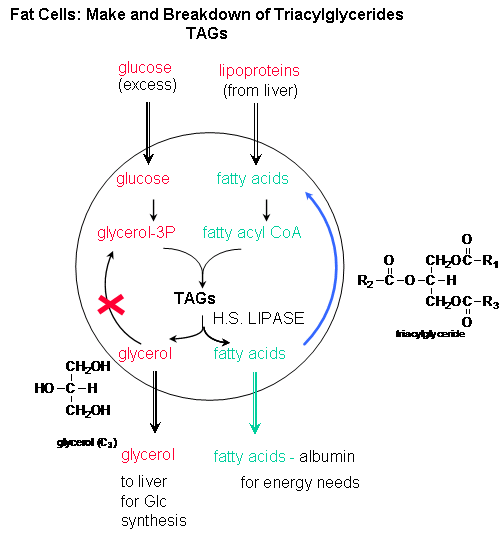
The net effect is that insulin promotes fat storage in fat tissue.
animations: diabetes from the American Diabetes Association
Type II Diabetes: Molecular Defects in Insulin production and signaling
Organ Profile: In this disease, insulin does not seem to be able to stimulate its normal effects. Pancreatic islet cells are defective in secreting normal insulin amounts. However, the major reason appears to be that cells become resistant to insulin. The resistance might occur through defects in the insulin receptor or any down stream factor in the signal transduction cascade initiated by insulin binding to its receptor. Some people don't need insulin pills since they have adequate levels. Other medications, such as those that block dietary glucose absorption in the small intestine, are useful. Dietary (decreased sugar consumption) and life style changes (increased exercise, etc) are useful in treating the disease. We will discuss normal insulin action below, but wait to discuss how insulin resistance (the hallmark of Type II Diabetes) develops until the unit on obesity.
Insulin resistance affects many organs. It inhibits glucose uptake into muscle and adipose cells, which causes blood levels of glucose to rise. Glucose gets into liver cells through a different transporter than the one affected by insulin in muscle and fat cells, so insulin resistance does not seem to affect glucose uptake into the liver. Insulin still binds to its receptor in the liver, but the net effect is a signal to the cells to increase glucose synthesis (through the gluconeogenic pathway), which the liver sends into the blood, further increasing blood glucose levels.
Given the amount of tissue involved, you would expect the muscle to remove most of the blood glucose when insulin binds to the muscle receptor. This organ does remove about 80% of the blood glucose. However if you get rid of insulin receptor in muscle using genetic techniques (obviously in a mouse model), no big changes in blood glucose occur, suggesting that the body adapts to this. If you remove the insulin receptor from fat tissue, it cant' store triglycerides anymore since glucose is needed in the fat cells for that as we saw in the figure above. If its removed from both tissues, insulin resistance results.
Insulin regulates triacylglyceride synthesis in fat cells. It does so by leading to glucose uptake. It also inhibits the enzyme which hydrolyzes fatty acids from triacylglycerides, hormone-sensitive lipase. If insulin is not present, or not working well, adipose cells cant' synthesize TAGs. Blood lipids increase. In other cells (like muscle), excess blood fats can lead to fatty acids uptake into the cell and storage of fat in muscle. This might also cause insulin resistance.
Insulin Signaling Anamolies: When insulin binds to its receptor, the receptor/kinase phosphorlylates itself, which then leads to the binding of other proteins to the activated receptor and their phosphorylation. One of these proteins leads to changes in gene transcription. The other, IRS1, a "scaffolding protein" leads to the movement of the GLUT4 protein (Glucose transport protein) to the cell surface. These activities are shown schematically in the figure below. IRS-1 binds PI 3-kinase (PI3K) that causes phosphorylation of a certain type of membrane lipid (a phosphatidyl inositol derivative). This phosphorylated lipid binds to and recruits to the membrane other proteins (PDK1 and Akt). The PDK1 becomes an active kinase, which phosphorylates and activates Akt. The active Akt, also a kinase, phosphorylates other proteins which ultimate leads to the movement of the GLUT4 protein to the membrane, where it can transport glucose into the cell.,
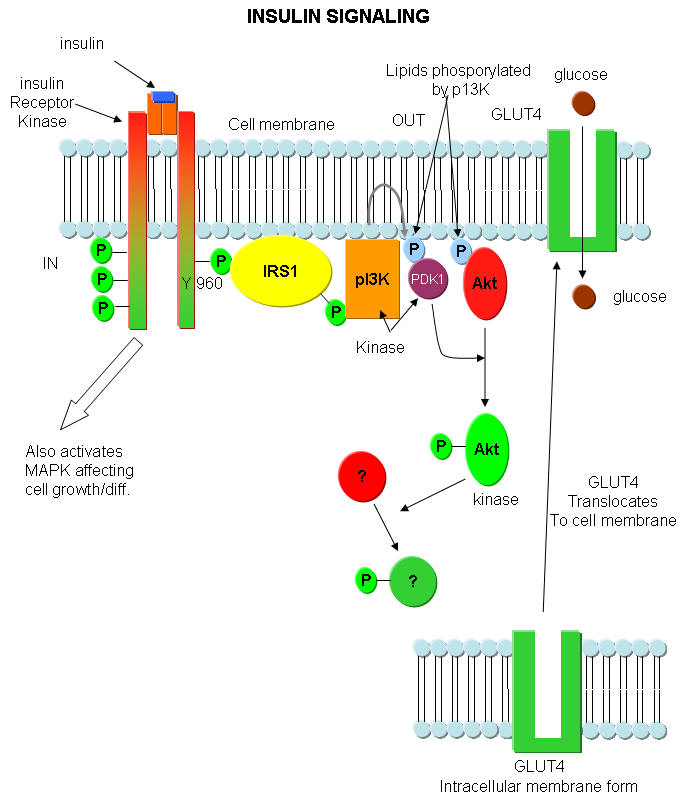
Figure above adapted from:http://www.biochemsoctrans.org/bst/031/1152/bst0311152.htm
Below is a graphic of
insulin signaling from Biocarta
:
http://www.biocarta.com/pathfiles/h_insulinPathway.asp, submitted by
Walter O'Dell, PhD which shows detail into the transcriptional effects of
insulin.
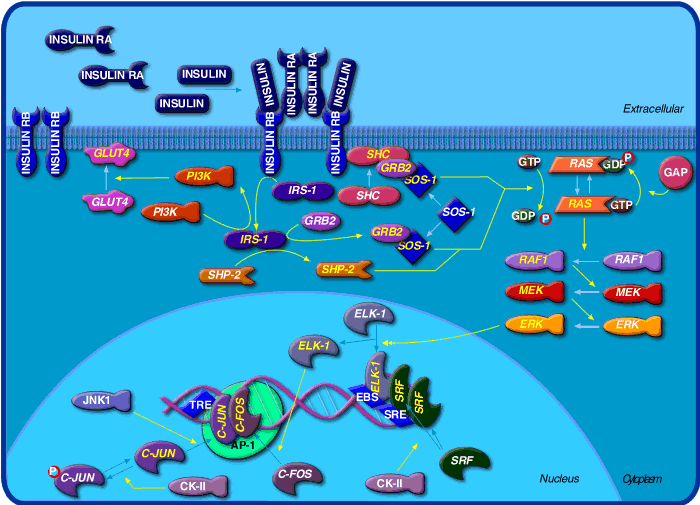
Something runs amok in this pathway in Type II diabetes. Obviously there are many stops where that could happen, from actual insulin binding to any of the downstream "effector" steps. We'll discuss some things that do happen in the section on Obesity.
Insulin effects on the liver: glucose and fatty acid metabolism
When we're awake and eating, we use glucose. When we're sleeping and fasting, we oxidizes fatty acids. The liver however has a special job: in the fasting state it must synthesis glucose since the brain needs it. So in the liver, gluconeogenesis is activated. How does the liver do both at one time: synthesize glucose and burn fat (which for the rest of the body would be at cross purposes)?
In the fasting state, the liver responds to the hormone glucagon and breaks down glycogen reserves to glucose, the same response we saw in muscle if we had to flee from a saber-toothed tiger (when epinephrine is released). It also activates gluconeogenesis. Both pathways give glucose. Instead of using it for its own energy by activating glycolysis, the liver sends the glucose into the blood where it can go to the brain, which needs a steady supply of glucose. Under the same conditions, the livers oxidizes fatty acids for its own energy supply.
In the feed state, the liver responds to insulin and stops gluconeogenesis, the synthesis of glucose, and also the oxidation of fatty acids. This makes sense: there is enough glucose around to use for energy. Why should the liver make more?
What happens in diabetes? The liver doesn't sense glucose availability and hence makes more through gluconeogenesis, which it then ships out into the blood, contributing to excess blood sugar levels. You would expect that the aberrant signaling that tells the liver to make more glucose in the diabetic state (insulin resistance) would also cause the liver to oxidize fatty acid. It turns out that that doesn't happen. The liver stops oxidizing fatty acids. It's as if it got the insulin signal of energy abundancy but applied it only to the breakdown of fatty acids. Instead of breaking them down, it actually uses fatty acids to synthesize triacylglycerides, which accumulate aberrantly in the liver. Insulin, through the insulin receptor, sends multiple signals into the cell through the kinase activity of the receptor. Transcription factors are activated and inhibited. One, that shuts down glucose synthesis (Foxo1) is not shut down appropriately. The other (Foxo2), is more sensitive to insulin, and gets shut down.
Why the pandemic?
To understand this, we first have to study obesity, which is closely linked to Type II diabetes. That's in the next unit.