04/05/2007
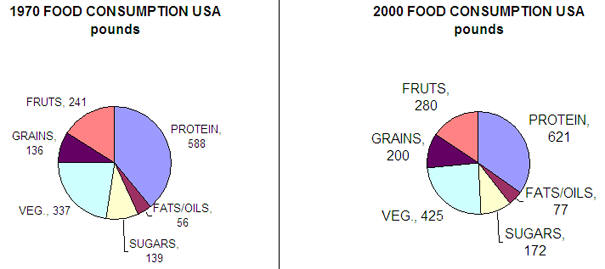
Its easier to address the question of why levels of obesity are growing faster than diabetes. We eat more food, exercise less, watch more TV, and surf the web. We drive and don't walk. Here are some data.
credit: National Geographic. August 2004, pg 49
Calories expended in half hour activity (unless noted) for 150 lb man
| Activity | Cal. | activity | Cal. |
| Email colleague (1 min) | 2 | Walk to colleague office (1 min) | 4 |
| Ride Elevator (2 min) | 3 | Take stairs (2 min) | 19 |
| Order take-out (1 min) | 1 | Cook Meal | 70 |
| Load dishwasher (10 min) | 23 | Wash dishes | 80 |
| Watch TV | 35 | Play cards | 52 |
| Go to car wash | 35 | Wash car at home | 104 |
| Play video game | 53 | Play basketball | 280 |
| Mow lawn/riding mower | 88 | Mow lawn/power mower | 193 |
credit: National Geographic. August 2004, pg 57
![]() WebCT:
Serving size
WebCT:
Serving size
BMI |BMI Calculator | Problems with BMI use alone
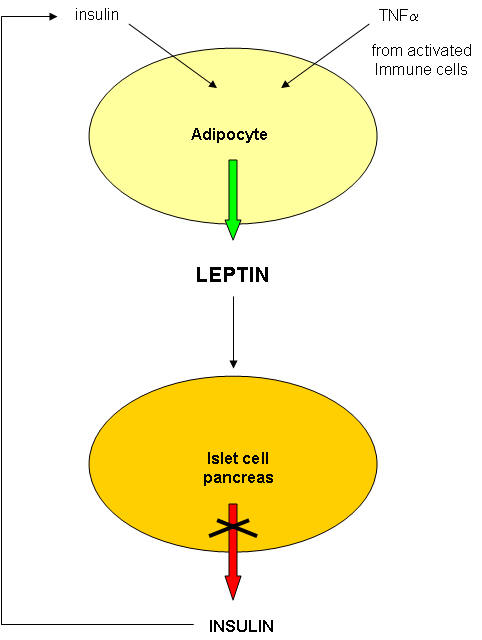
The molecular bases of obesity are extremely complicated (i.e. worse than anything we've seen this semester.) In addition, obese people usually have diabetes (type II). We saw how carbohydrate and fat metabolism are intertwined in the fat tissue. In addition, we saw how insulin has different effects on the muscle, adipose cells, and the liver. Multiple organs with different roles are involved in the pathogenesis of diabetes.
Diabetes probably does not cause obesity. Type I diabetics, which lack insulin, are often thin. However, obesity, through its systemic affects throughout the body, does appear to lead to type II diabetes.
So where should we start to understand obesity and its role in promoting type II diabetes? It makes sense to first look at the biochemical properties of fat tissue, composed of fat cells or adipocytes, to get clues as to the nature and causes of obesity. Fat in the form of triacylglycerides can be abnormally deposited in muscle and liver in obese and diabetic individuals as well, but we will only consider here fat stored in adipose cells. If adipose cells play a role in diabetes, they must do so by affecting other organs.
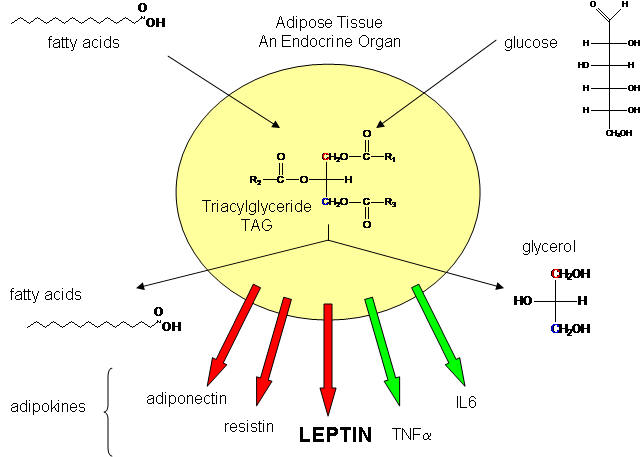
Not so long ago, adipose tissue was viewed as a passive storage tissue for nonpolar fat molecules (triacylglycerides - TAG). It's main role was to bring in glucose and fatty acids to synthesize the TAGs, or break it down and release fatty acids and glycerol, under the control of the hormone glucagon. Now we realize that fat cells play a more fundamental role in both the intake and storage of energy molecules by receiving and sending out protein signals like the immune cells we have studied. Signaling proteins released by immune cells were called cytokines. Signaling proteins released by fat tissue, acting as an endocrine organ, are called adipokines. They include leptin, resistin, adiponectin, IL-6 and TNFα, as shown below. We will discuss two in some detail:
leptin (form the Greek leptos meaning light)
TNF-α - tumor necrosis factor .
In addition, there are many proteins that stay inside the fat cell (i.e. they are not secreted). These include proteins involved in lipid metabolism and the expression of those proteins from their genes. We will discuss in some detail two proteins that act as transcription factors in fat cells:
NF-κB
PPAR-γ.
Fat cells in the body, which take up only about 10% of the total insulin-regulated glucose taken up throughout the body, helps regulates glucose metabolism throughout the body. It does so, in part, by concerted action of these four proteins (and many others as well).
1. Leptin: This protein, discovered in 1994, appears to the main regulator of the energy storage balance in the body. It is secreted by adipocytes and binds to receptors in the hypothalmus of the brain, and also on T cells (suggesting a link between fat tissue and the immune system). Its structure is shown below, followed by a table which show how blood leptin levels vary under different states.
![]() Chime Structure:
Human Leptin
Chime Structure:
Human Leptin ![]()
| Condition | Leptin Levels | Biological Effect |
| ↑ Body Fat | ↑ Leptin | ↓ food intake |
| ↓ Body Fat | ↓ Leptin | ↑ food intake AND ↓ energy expenditure |
| mutant Leptin | ? | ↑↑↑ obesity |
| Obesity | ↑ Leptin | would expect to ↓ food
intake and lose weight but obese become leptin resistant |
Summary of Leptin Effects
Increased leptin signals increased fat storage, which should lead to decreased food intake
Decreased leptin signals decreased fat storage (during lack of food), which should induce increased food intake (by increasing desire - unconscious, conscious) and decreased energy expenditure (lower metabolic rate so you don't burn as many calories as usual since normal fat energy reserves are depleted).
Obese become leptin resistant - and don't respond to leptin if given as drug
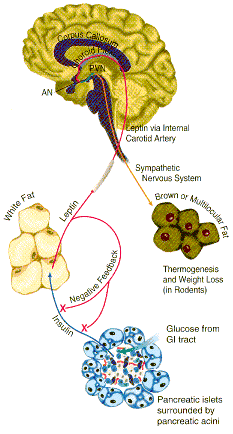
Leptin binds to the leptin receptor on neurons in the hypthalmus of the brain. This leads to decreased levels of a brain neuropeptide Y, which decreases appetite and signals fat cells to break down triglycerides, release fatty acids, which go to other tissues for oxidation. This decreases fat deposits.
What signals fat cells to release leptin?
credit: http://www.rndsystems.com/images/mini/r_leptin_bg.gif
That the body has such an elaborate system to control food intake and weight makes great sense from an evolutionary point of view. Our earlier ancestors, presumably hunter-gathers, found food scarce. Those that could, in the presence of food, best store extra ingested food in the form of fat, would have an advantage over those that could not. These people would have had what have been called "thrifty genes", which would promote fat storage in time of plenty (presumably few and far between). In a modern society when food is always available, we eat three large meals a day or more, when meals have become super-sized, and we engage in little physical activity, these same "thrifty" genes, which were useful for our ancestors, are detrimental to our health. We store excess calories and fat, and are increasingly becoming obese. Decreased leptin levels starts the "thrifty" response of our bodies.
2. TNF-α : TNF is the main inflammatory cytokines secreted by macrophages and is also released by fat cells. One major target of TNF is adipocytes themselves, where it inhibits transcription of some genes and activitates expression of others.
Decreased expression (i.e. inhibition) of the following genes occurs in adipocytes treated with TNF-α:
Longer treatment of cells leads to decreased levels of the following proteins:
All of these effects would lead to insulin resistance (i.e. insulin would appear not to work). That this inflammatory mediator is secreted from both fat cells and macrophages and has such an effect on glucose levels suggests that obesity represents a chronic state of inflammation which has systemic consequences throughout the body.
3. NF-κB: It appears that many of the genes whose transcription are inhibited by TNF require the activation of a specific protein transcription factor in fat cells called NF-κB. Drugs are available that inhibit NF-κB. If given to adipocytes after TNF stimulation, the effects of TNF are NOT observed. This suggests that the effects of TNF are mediated through NF-κB. This protein is usually bound to a protein inhibitor, IF-κB, which prevents it binding to DNA and acting as a transcription factor. In the presence of TNF-α, IF-κB becomes free from NF-κB, and now, in its active form, can act as a transcription factor. The release of NF-κB from its inhbitor, IF-κB, and its subsequent binding to DNA, is shown below.
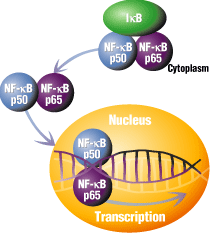
credit: http://www.piercenet.com/Proteomics/browse.cfm?fldID=843944B3-65F9-42A9-BF7A-6F91B4A6FBBB
The figure below shows how TNF might liberate NF-κB from its inhbitor, IF-κB.

credit: Biocarta : http://www.biocarta.com/pathfiles/h_stressPathway.asp submitted by Eric Schaefer, PhD,
4. PPAR-γ: Some genes inhibited by TNF (through activation of NF-κB) require another transcription factor, PPAR-γ, or peroxisome proliferator activated receptor for their normal expression. PPARs appears to play a significant role in the differentiation of cells into fat cells, in the metabolic control of fat metabolism, and have been implicated in the development of insulin resistance.
PPARs (α found in the liver and γ in fat cells) bind fatty acids, their metabolites, and various drugs, which on binding lead to shape changes and binding/unbinding of PPAR from its response element, PPAR response element, that controls expressions of many genes involved in lipid metabolism. They are lipid sensor transcription factors. Drugs that bind and activate PPAR-γ help sensitize cells to insulin. Drugs that bind and activate PPAR-α help lower blood lipid levels.
PPAR-γ (expressed in fat cells) has the key role in the control of differentiation of precursors of fat cells into adipose cells. If the PPAR-γ is expressed in non-adipocyte cells, they become adipocytes. If the biological activity of PPAR-γ in fat cells is activated (by drugs for example), then fat cells become better storage places for fats. This might lead to the movement of abnormal lipid deposits from muscle and liver to the fat cell, and a reduction in blood triacylglycerides, conditions which would be beneficial to a diabetic. (PPAR-γ turns on genes necessary for fat storage, including glycerol kinase, lipoprotein lipase, and fatty acid transporters). So if TNF, through activation of NF-kB, inhibits gene expression mediated by PPAR-γ, then diabetes might result. TAGs would be broken down, and blood levels of fatty acids would increase).
Another effect of activation of PPAR-γ by drugs is changes in adipokine expression and secretion which would have a systemic effect throughout the body. These changes decreases insulin resistance. TNF levels decrease, which would promote insulin sensitivity, and counter resistance. Also adiponectin secretion increases. This increases glucose utilization by muscle and decreased synthesis by liver, leading to lower blood glucose levels. The main effect of the drugs are on PPARs in fat cells, which leads to systemic responses.
PPAR-α is expressed in the liver. In the fasting state (or during sleep), the liver imports fatty acids from fat cells. This turns on PPAR-α activity, which leads to fatty acid oxidation. This ultimately decreases blood triacylglycerides (from decreased liver synthesis), which helps cardiovascular function.
Let's get back to the inhibition by TNF of genes regulated by PPAR-γ. How can you get inhibition of gene transcription from PPAR-γ induced genes when TNF activates a transcription factor NF-κB? The simplest explanation is that one transcription factor NF-κB prevents the other, PPAR, from binding and or activating gene transcription from the other. No one knows for sure. First more information on PPARs.
|
"PPARs are an important class of nuclear proteins
involved in diabetes, obesity, Credit: http://www.bmb.leeds.ac.uk/illingworth/ppar.htm PPAR α (468 amino acids, chromosome 22) activates many lipid oxidation genes. It also promotes gluconeogenesis and has anti-inflammatory actions. PPAR α is expressed mainly in brown fat and liver, followed by heart, kidney and gut. It is the target for fibrate drugs, such as gemfibrozil [which lower blood lipids]. PPAR γ (475 / 505 amino acids, chromosome 3) is necessary and sufficient for adipocyte differentiation. It is expressed mainly in adipocytes and in the foam cells of atherosclerotic lesions. It is the target for thiazolidinedione drugs [insulin sensitizing agents], such as rosiglitazone [and troglitazone (TGZ)] PPAR γ agonists [drugs that bind to PPAR γ and activate it, like troglitazone (TGZ)] increase insulin sensitivity, "normalize" serum lipids and suppress the synthesis of leptin, resistin and TNF α by fat cells (but probably not by macrophages)." Peroxisome proliferators (PPs) are small molecules that increase [liver] peroxisomes (an organelle in cells involved in lipid metabolism). PPs include plasticizers, herbicides, unsaturated lipids, eicosanoids [prostaglandin family], fibrates [plasma lipid lowering drugs] and thiazolidinediones [insulin sensitizing agents like troglitazone]. PPs bind to peroxisome proliferator activated receptors (PPARs) which are nuclear proteins that modify gene expression. |
Here are the net effects:
When PPAR-γ is expressed in cells, it appears to inhibit NF-kB mediated gene expression. Adding the drug troglitazone to fat cells, which binds to PPAR-γ and stimulates its activity) stops NF-kB mediated decreases in gene expression. This reverses the effect of the inflammatory cytokine TNF on fat cells (which activates NF-kB). This promotes insulin sensitivity (good) and reverses insulin resistance. The drug does not stop NF-kB activation or its binding to DNA, just the functional activity (activation or inhibition of gene transcription) of the bound NF-kB.
When NF-kB is activated by TNF or other means, it inhibited PPAR mediated gene expression, even though NF-kB does not bind in the promoter or response element area of the PPAR-γ gene.
NFkB and PPAR seem to mutually regulate each other. Hence, if you could inhibit NF-kB activity you would promote PPAR activity, and minimize insulin resistance (i.e. improve insulin sensitivity). This appears to be the way in which certain drugs that promote PPAR activity (PPAR agonists like troglitazone - TGZ) improve insulin sensitivity. The drugs block NF-kB dependent inhibition of transcription. However, NF-kB can still bind to target sequences on the D DNA. All of this suggests some intermediary molecule that interacts with both PPAR-γ and NF-kB. (See J Biol Chem 278 (30): 28181-28192. 2003 for much more detail).
We saw previously that inflammation can lead to insulin resistance, a hallmark of Type II diabetes. Now it appears that obesity turns on the inflammatory response of the innate immune system. Some speculate that these biochemical responses lead to increased glucose availability to the brain at the expense of the peripheral organs.
New 4/18: Evidence shows that diabetes does not cause obesity, but the obesity, through the mechanisms above (TNF affects, etc). can lead to diabetes. There seems however to be a logical conundrum. If you give a type II diabetic drugs that activate PPAR-γ (troglitazone - TGZ, for example) their fat cells will start synthesizing TAG (possibly leading to the removal of fat from muscle and liver to the fat cell) which diminish insulin resistance and increase insulin sensitivity, which is what you want to do in the treatment of type II diabetics. However, this increased storage of fat in adipose cells increases fat tissue and causes weight gain (in part from increased food consumption). But isn't increased fat viewed as an inflammatory state which causes fat cells to release more TNF, which promotes insulin resistance - i.e. type II diabetes? Is there a way out of this dilemma? Some PPAR-g activation effects might be short term (which would be beneficial in the treatment of type II diabetes. However others (eventual longer term weight gain) may be detrimental.
Summary of other adipokines:
Adipoectin (Adipocyte complement-related protein of 30 kDa - Acrp30): This protein can bind to muscles cells and promote the breakdown and oxidation of both carbohydrates AND fats. Blood levels of this protein decrease in diabetes and obese people.
Relaxin:
By what mechanisms does obesity lead to diabetes? There must be some connections between the signaling pathways involved in the diseases. New data suggests that an organelle called the endoplasmic reticulum (ER) is involved. The ER is ia long, tube-like structure involved in protein synthesis, folding, and secretion from the cell. Some proteins that are synthesized in the cytoplasm are destined for insertion into various membranes in the cell, or secretion from the cell. These subclasses of proteins are translocated across the ER membrane as they are synthesized. While in the ER, sugar residues are covalently attached to the protein, before the protein is delivered to its final destination (a membrane or the outside of the cell).
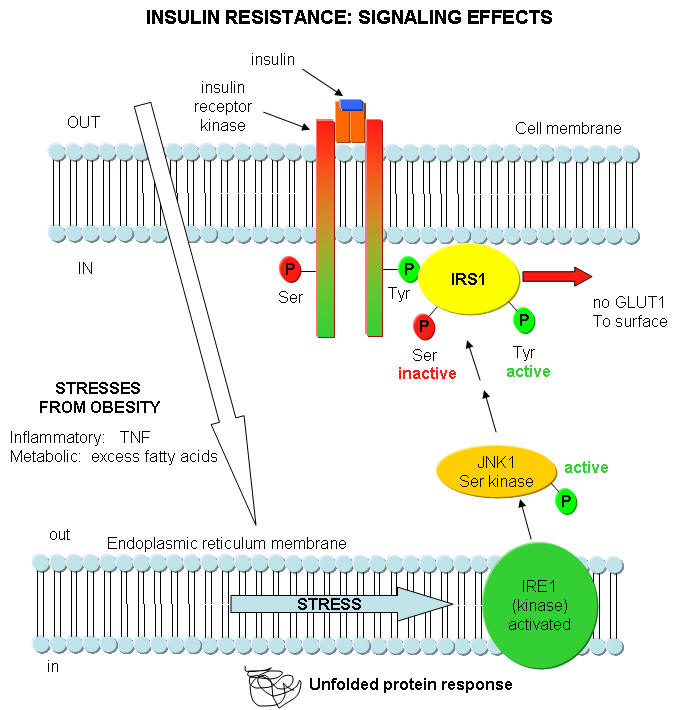
What happens in Type 2 diabetes that decreases the activity of insulin? Once again, insulin starts its activity by binding to its receptor, leading to the activation of the insulin receptor tyrosine protein kinase, which phosphorylates itself and other proteins in the cell, including insulin receptor substrate 1 (IRS-1). As we mentioned throughout this course, for every event the "turns on" a biological activity, there must be one that attenuates that activity. One way that insulin effects are decreased is through the activation of another protein kinase, JNK1, that phosphorylates another amino acid, serine, in proteins. JNK1, a serine protein kinase, stands for Jun-amino terminal kinase. What activates JNK1? It turns out that fatty acids (much you might expect to be elevated in obese people) and immune system signaling molecules that we studied before: tumor necrosis factor (TNF). TNF is released from macrophages and is one of the main mediators of inflammation. JNK1 activity is increased in liver, muscle, and fat cells in obese people. These are the same tissues that insulin affects. This suggests a mechanism for a decreased insulin response in the obese state, through the activation of the serine protein kinase JNK1 by fatty acids and inflammatory cytokines. You can actually delete the JNK1 gene in mice prone to diabetes. The mutants do not show insulin resistance.
Where does the ER come into play? A new idea is that ER stress caused by a variety of stimuli contributes to insulin resistance by activating JNK1. The stress could be related to the immune system (from chronic inflammation as from TNF) and from metabolic dysregulation (leading to increased fatty acid). Many events can cause ER stress that could lead to protein folding problems. Examples include lack of nutrients, elevated temperatures, viral or bacterial infections, lack of oxygen, etc. These cause metabolic "strain" on the system, leading to increasing problems in protein folding in the ER. They all result in increased levels of unfolded proteins in the ER. As this itself is a stress, the cells activates another pathway, the unfolded protein response (UPR). In this process, misfolded and unfolded proteins are degraded. Studies show that the unfolded protein response leads to increase in JNK1 activity and increased phosphorylation of Ser residues in insulin response substrate 1 (IRS-1). All this is consistent with chronic immune and metabolic stress, both enhanced by obesity, decreasing insulin signaling, leading to insulin resistance.