06/21/2006
Before a protein or other biological macromolecule can be rigorously studied from a structural and functional basis, it must be purified. The problems that can arise during protein purification become clear when one considers that a single protein has to be purified from a mixture of as many 10,000 proteins, each of which are made up of the same constituent amino acids. Proteins differ in size (how many amino acids), charge (how many positively and negatively charged amino acids), and in sequence and presence of specific binding sites on the proteins. Any technique that could be used to purify protein must be based on these inherent differences. Three major column chromatography techniques used to separate proteins (or other macromolecules) will be discussed below.
TYPES OF CHROMATOGRAPHY
1. Separation on the basis of size- Gel Filtration chromatography:
Proteins of different sizes are separated on a column in which the stationary phase is an polymerized agarose or acrylamide bead, which contain pores of various sizes. A smaller protein in the mobile phase (aqueous buffered solution) may enter the pores in the bead, while a larger protein may not, due to size restriction. The result is that a larger fraction of the overall volume of the column is available to the smaller protein, which thus spends a longer time on the column and is eluted by the mobile solvent after the larger protein. The shape of the protein (not only its size) also determines whether the protein will enter the pores. We used this technique to separate dye encapsulated in liposomes from free dye in Lab 1.
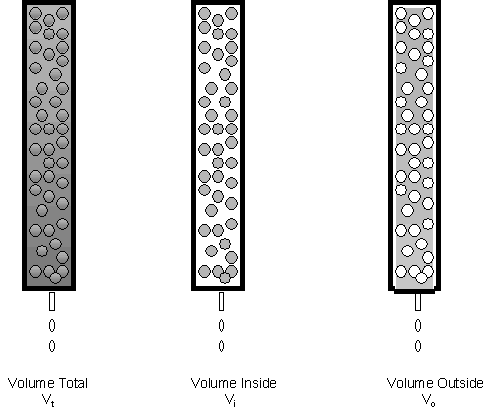
Lets consider in more depth the theory of gel filtration chromatography. Several different column volumes can be defined as shown below.
If we consider the mass of the beads to offer a negligible amount to the volume of the bead, the actual volume in the bead represents trapped solution and the "stationary" phase. The volume around the bead is called the void volume, Vo. Also, Vi = Vt - Vo.
A solute elutes from the column in a broad peak. If the sample volume applied to the column is very small compared to Vt, the volume at which a solute elutes, Ve, is considered to be the center of the elution peak. This is true when Vsample << Ve.
If we view this chromatography has a partitioning of solute between the mobile and stationary phases, we might be interested in what fraction of the stationary phase, Vi, a solute might partition into. Such a ratio would be given by:
K = (Ve-Vo)/Vt-Vo)
where Vt-Vo (=Vinside) represents 100% of the stationary phase, where K is a distribution coefficient. Consider two cases:
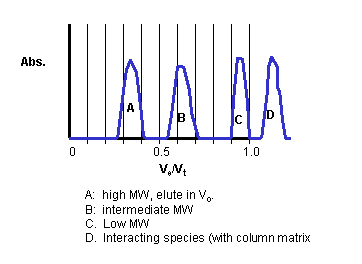
Hence K is a partition coefficient, which varies from 0 - 1, and represents that fraction of Vi into which a solute could partition. This K is not exactly a partition coefficient, however, since the actual volume of the gel matrix is assumed to be zero above. The graph below shows typical Ve as a fraction of Vt for solutes of different size (x axis is Ve/Vt).
Size and shape relationships:
K depends on the size and shape of the solute. The size and shape of an object determines is flow properties in a fluid. Frictional resistance (a force which has the opposite direction as the velocity, another vector quantity, can be shown to be proportional to the velocity.
Ff α v or Ff = - fv
where f is the frictional coefficient, which depends on shape. Clearly, the bigger the object, the more frictional resistance to movement. For a sphere it can be shown that:
f = 6πηRs
where η is the viscosity (measure of the resistance to flow of a liquid - water has a low viscosity, real maple syrup a high viscosity), and Rs (Stokes radius) is the radius of the hydrated sphere (the larger Rs, the larger the frictional coefficient, the larger the Ff which resists motion). For an irregularly shaped object , the Stokes radius is the radius of a sphere that would have the same frictional coefficient as the object. Hence the Rs for a protein molecule that was not spherical in shape would be much larger than the Rs for another protein molecule of identical molecular weight that was spherical. Hence the Ve and the K value for a solute on a gel filtration column would best be related to the Stokes radius, since Rs values takes into account both size and shape.
It can be shown that:
Rs α erf-1 (1-K) or Rs = A + B erf-1 (1-K)
What would a plot of Rs vs erf-1 (1-K) look like?
For globular proteins, you can also show that
K = -A log (MW) + B or more rigorously
K = -A' log Rs + B'
log Rs α log MW of the protein, and that, more practically,
a plot of log MW vs either K or Ve is linear with a negative slope.
Hence gel filtration can be used to determine the MW of an unknown spherical (globular) proteins when compared to a standard curve generated used other globular proteins of known MW. In Lab 1 we separated liposomes-encapsulated dye from free dye. That separation was trivial since it was equivalent in difficult to separating elephants from mosquitoes. It is much more difficult to separate proteins of similar size, a problem often faced in the lab.
2. Separation on the basis of charge- Ion exchange and chromatofocusing chromatography:
The chromatography resin in this type of chromatography consist of an agarose, acrylamide, or cellulose resin or bead which is derivatized to contain covalently linked positively or negatively charged groups. Proteins in the mobile phase will bind through electrostatic interactions to the charged group on the column. In a mixture of proteins, positively charged proteins will bind to a resin containing negatively charged groups, like the carboxymethyl group, CM (-OCH2COO-) or sulfopropyl, SP, (-OCH2CH2CH2SO3-) while the negatively charged proteins will pass through the column. The positively charged proteins can be eluted from the column with a mobile phase containing either a gradient of increasing salt concentration or a single higher salt concentration (isocratic elution). The most positively charged protein will be eluted last, at the highest salt concentration. Likewise, negatively charged proteins will bind to a resin containing positively charged groups, like the diethylaminoethyl group, DEAE (-OCH2CH2NH(C2H5)2+) or a quarternary ethyl amino group, QAE, and can be separated in an analogous fashion. In chromatofocusing, the mobile phase contains a gradient in pH, and bound proteins are eluted when the pH reaches the isoelectric point of the protein, at which the proteins have no net charge. In this lab, you will use QAE and SP Sephadex ion exchangers.
3. Separation on the basis of specific binding sites on the protein- Affinity Chromatography:
In this technique, the chromatography resin is derivatized with a groups which will bind to a specific site on a given protein of interest. It may be a group which binds to the active site of an enzyme (such as benzamidine-agarose which is used for the purification of trypsin) or an antibody which recognizes a specific amino acid sequence on a protein. For example, an antibody can be made to a specific peptide from albumin, the antibody covalently linked to agarose, and the antibody-agarose column then used to purify albumin specifically. This is a powerful technique, since antibodies can theoretically be made that will bind selectively to any given protein. Knowing only the DNA sequence of a protein which has never been previously isolated, the following scheme could be used to purify the unknown protein. The amino acid sequence of the unknown protein can be derived from the DNA sequence. A 10-12 amino acid peptide from that protein can be synthesized in the lab, and an antibody raised against the peptide. The antibody will most likely bind to the unknown protein as well as to the peptide, and hence could be used to purify the protein.
4. Hydrophobic Interaction Chromatography (HIC)
5. Reverse Phase Chromatography
6. TLC