04/23/2006
In the previous lab, we discussed the motion of a particle in a fluid medium, where the sedimenting force was the gravitational attraction (on a flowing liquid), and the opposing force was a frictional force proportional to the velocity of the particle. These forces act in opposite directions and eventually balance each other, leading to the uniform motion of the particle in the mobile liquid medium (i.e. the particle moves at constant velocity). If the external field is an electric field instead of a gravitational field, there are two ways which a macromolecule will respond to the external field. If the molecule is charged, it will migrate in an electric field to the electrode of opposite charge. This is the principle underlying the technique of electrophoresis. If the macromolecule has an asymmetric distribution of charge (i.e. has a permanent dipole moment), the molecule will tend to orient in an electric field. This principle provides the basis for the techniques of electric birefringence and dichroism. We will discuss only electrophoresis.
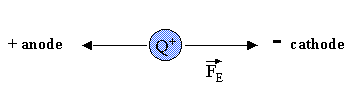
Consider the simple case of a charged particle (+Q) moving in an electric field (E) in a nonconducting medium, such as water. If the particle is moving at a constant velocity toward the cathode (- electrode), the net force Ftot on the particle is 0 (since F=ma, and the acceleration (a) of the particle is 0 at constant velocity). Two forces are exerted on the particle, one FE, the force exerted on the charged particle by the field, which is in the direction of the motion (toward the cathode), and the other, Ff, the frictional force on the charged particle, which retards its motion toward the cathode, and hence is in the direction opposite to the motion (toward the anode (+) electrode). This is shown in the diagram below:
Therefore:
(1) Ftot = Fe + Ff = 0,
where
(2) Fe = QE (the electric force) and
(3) Ff = -fv (the frictional force),
where v is the velocity of the particle, and f is a constant called the frictional
coefficient. Equation (3) shows that the force Ff hindering motion toward the
cathode is proportional to the velocity of the particle. This is intuitive since one would
expect the higher the velocity the greater the Ff which would hinder the
motion. The frictional coefficient depends on the size and shape of the molecule. The
larger the molecule, the larger the frictional coefficient (i.e. more resistance to motion
of the molecule). It can be shown that the frictional coefficient for a spherical particle
is given by
(4) f = 6πηRs,
where η is the
viscosity (measure of the resistance to flow of a liquid - water has a low viscosity, real
maple syrup a high viscosity), and Rs (Stokes radius) is the radius of the
hydrated sphere (the larger Rs, the larger the frictional coefficient, the
larger the Ff which resists motion toward the cathode). From (1), (2), and (3),
Fe = Ff , or
(5) QE = fv.
Hence v/E = Q/f = U = electrophoretic mobility, or
(6) U = v/E = Q/6πηRs.
Therefore, the electrophoretic mobility U is
proportional to the charge density (charge/size, Q/Rs) of the particle.
Macromolecules of different charge density can thus be separated by electrophoresis. This
discussion deals with the simplest case, since in reality there are counter ions in the
solution (from salts) which would form a cloud around the charged macromolecule, and
partially shield the charged particle from the electric field E.
Modern day electrophoresis is conducted in solid gels (such as polyacrylamide), which are formed from liquid acrylamide solutions after the addition of a polymerizing agent. The solid gel is porous to solute and solvent molecules and serves as a medium for electrophoresis while helping to eliminate convection forces in the liquid which would interfere with the separation. Electrophoretic experiment have been conducted on the space shuttle in weightless conditions in order to prevent such perturbations.
One complication that affects this idealized description of electrophoresis in polyacrylamide gels is that the gels have pores through which the macromolecules move. As in gel chromatography, the smaller molecules can pass through the pores more readily than larger molecules, so there is an additional sieving mechanism that contributes to the effective mobility (Also, the gel could alter the local effective electric field). The sieving effect of the gel actually increases the resolving power of this techniques.
It has been determined that the actual electrophoretic
mobility of the protein, U, is a function of the mobility of the protein in a concentrated
sucrose solution (Uo) and T, the total concentration of the acrylamide in the polymerized
gel. The higher the concentration of acrylamide in the unpolymerized gel solution, the
smaller the size of the pores in the polymerized gel. An equation showing the relationship
between U, Uo, and T is shown below:
log U = log Uo - KrT,
where Kr is the slope of a plot of log U vs T for a given protein. Since Kr
is a function of the radius of the molecule, it is possible to determine the molecular
weight of a protein molecule by performing several electrophoretic separations in gels of
different acrylamide concentrations (T), and extrapolating results to T = 0, hence
eliminating pore size effects. Problems arise, however, if the proteins are not spheroid
in shape
Is there any way to obtain molecular weight information, in addition to purity determination, on a single gel?. What would result if two different proteins, each with the same molecular weight and total net charge, but different shapes, were run on a single acrylamide gel? The one having the more elongated shape (large Stokes radius) would have a lower electrophoretic mobility (U = Q/6πηRs). A larger Rs would also cause the protein to enter the pores at a slower rate. Hence both electrophoretic mobility and sieving effects would cause this protein to run anomalously slow and have a higher apparent molecular weight. Also imagine two globular proteins of different size but with compensatory charge differences which might allow the two proteins to migrate at the same speed in the gel.
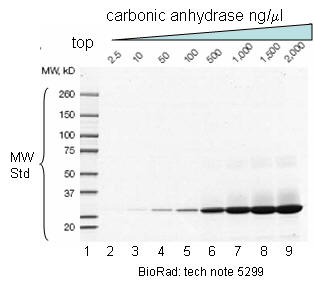
A common technique used to simplify the interpretation of electrophoretic runs in a single gel is to run the gel under denaturing conditions. The denaturant of choice is usually sodium dodecyl sulfate (SDS), which is an ionic detergent with the structure CH3(CH2)10CH2OSO3- (a single chain amphiphile). This detergent binds to and denatures most proteins, with about 1.4 g SDS binding/g of protein (about 1 SDS/2 amino acids). Since there is 1 negative charge/SDS, the binding of SDS masks any of the charges on the protein, and gives all proteins an overall large negative charge. Additionally, SDS-proteins complexes have been shown to generally have a elongated cylindrical-like shape. Since the amount of SDS bound per unit mass of protein is constant, the overall charge density on all proteins is similar, so the electrophoretic mobility is only determined by sieving effects. SDS also eliminates shape differences in the proteins as a variable which determines sieving, since all proteins have the same general rod-like shape. (The use of SDS is analogous to the use of 8M urea in the gel chromatographic separation of proteins to determine molecular weights). Mobility becomes only a a function of the molecular weight of the protein, and not shape. The molecular weight of an unknown protein can be determined by comparing the protein's position on an SDS polyacrylamide gel with a series of known molecular weight standards from which a linear plot of the ln Mr vs Rf can be used to calculate unknown molecular weights. This is similar to the analysis in gel chromatography, where ln Mr is a linear function of Kavg, the distribution coefficient, when the gel is run under denaturing conditions. However, some proteins run anomalously on such gels (due to incomplete or excess binding SDS), so alternative techniques of molecular weight determination should be used in conjunction with this technique.
Proteins are usually heated in SDS to 100oC for 3 minutes, in the presence of a reducing agent such as b-mercaptoethanol, to completely denature the protein to a rod-shaped protein. Apparent molecular weight can be obtained under non-reducing conditions (without b-ME), but these should be considered just estimates. Running proteins both in the presence and absence of the reducing agent can provide important information on the subunit structure of a protein. A multimeric protein whose subunits are held together by disulfide bonds can be resolved into its individual components when the reducing agent is added. If the subunits are held together by noncovalent intermolecular attractions, the proteins will run identically under the denaturing conditions (SDS), which will eliminate subunit interactions, in the presence or absence of b-ME. To determine the subunit composition of a protein held together by noncovalent interactions, the electrophoresis should be performed in the absence of denaturing agents.
Note: Electrode nomenclature might be confusing to some of you. As mentioned above, cations move towards the cathode (where reduction occurs), so the cathode must be negative. Likewise, anion move towards the anode (where oxidation occurs), so the anode must be positive. This is opposite of what you might remember from General or Analytical Chemistry, when you discussed primarily galvanic cells. In galvanic cells, an electrical current is generated from a spontaneous set of redox half-reactions. We are dealing with electrolytic cells, such as in the electrolysis of water (2H2O(l) --> 2H2 (g) + O2(g)) or in the productions of Cl2(g) and Mg(s) from the aqueous electrolyte MgCl2(aq). In electrolytic cells, an power supply must supply the current to drive the nonspontaneous (unfavored thermodynamically) reactions, such as outlined above. Check out the review figure below.
TECHNIQUES
In this lab you will mount a SDS-polyacrylamide gel, apply proteins chromatographically separated in the previous lab, and perform the electrophoresis. The results from this lab should help you decide which proteins eluted in the different fractions from the G50 and ion exchange experiments. In addition, you should be able to determine the subunit composition of the proteins.
Polymerizing and pouring the gel:
Electrophoresis is performed in a porous, yet solid medium, to eliminate any problems associated with convection currents. Such media are formed from the polymerization of a liquid solution of agarose (used mostly for electrophoresis of DNA fragments and very large proteins) or acrylamide. Polymerization of acrylamide is initiated by the additions of ammonium persulfate in the presence of tetramethylenediamine (TEMED), along with a dimer of acrylamide (N,N'-methylene-bis(acrylamide) connected covalently between the amide nitrogens of the acrylamides by a methylene group. The structures of these compounds is shown below:
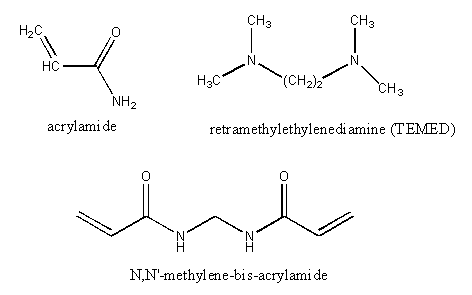
The free radical polymerization of the acrylamide is initiated on the addition of ammonium persulfate, which on dissolving in water, forms free radicals, as shown below:
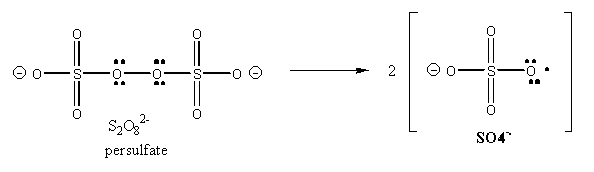
The radical initiate polymerization of the acrylamide, as shown below. The TEMED, through its ability to exist as a free radical, acts as an additional catalyst for the polymerization. A rigid gel is only formed, however, when N,N'-methylene-bis(acrylamide is added to the mixture during the polymerization, which cross-links adjacent acrylamide polymers as shown below:
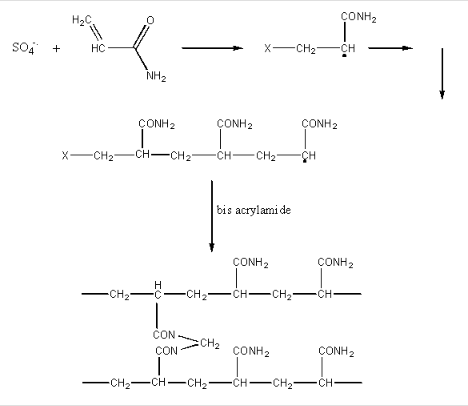
The amount of bis added during polymerization controls the degree of cross-linking, and hence the pore size of the polymerized gel. The effect of pore size is OPPOSITE to that in gel chromatography. In both cases, large proteins have a difficult time entering the pore. In gel chromatography, large proteins partition preferentially into the mobile liquid phase (the void volume) and are eluted most QUICKLY from the column. In electrophoresis, large proteins, which can not readily enter the pores in the gel, are not as easily transported by the electric field through the gel, and elute most SLOWLY. Pore size can not be controlled as accurately as in the manufacture of gel chromatography resins.
How do proteins migrate through the gel? A viscous protein solution is layered on the top of the gel in a small well molded into the gel during the polymerization process. The bottom and top parts of the gel are inserted into reservoirs containing a buffered solution and the appropriate electrode. The electric field is applied and the proteins migrate through the hydrated gel. The nature of the buffer solution in the reservoir and in the polymerized gel is important. The components of the buffer must not bind to the proteins to be separated. Additionally, the pH of the medium must be such that the proteins have the appropriate charge, so they will migrate in the expected direction.
Discontinuous gel electrophoresis:
There are many variations of electrophoresis commonly used. Gels can be polymerized in tubes, or slabs, and in the presence or absence of denaturing agents. Additionally, a given slab might consist of two separate slabs polymerized one on top of each other, each with a different acrylamide concentration and pH. This type is called discontinuous pH gel electrophoresis, or disc-electrophoresis. We will use this technique in today's lab. The two different pH and acrylamide concentration gels (the stacking gel, and running gel) are shown in the picture below.
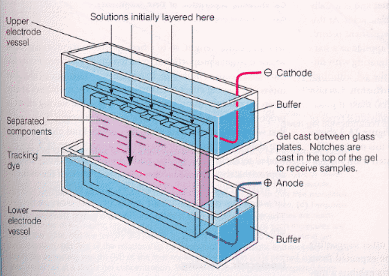
The stacking gel is a low concentration acrylamide (2-4%) polymerized in a Tris HCl buffer solution (pH 6.5) two pH units below that used in the running gel and the bottom reservoir (Tris HCl buffer, pH 8.7). The lower or running gel concentration varies from 7-15% acrylamide, depending on the molecular weight of the proteins to be separated. The upper buffer reservoir contains Tris buffered with a weak acid such as glycine (pKa2 = 9.6) to the same pH as the running gel.
What are the advantages in resolution of a discontinuous pH gel system? The main advantage is that the proteins electrophorese quickly through the stacking gel and "stack" at the interface between the two gels, before they enter the gels. This increases the compactness of the proteins before they enter the running gel and increases resolution. How does this stacking process work? When the electrophoresis is started, glycine ions from the upper reservoir (at pH 8.7) enter the stacking gel since at that pH they have a average partial negative charge. The stacking gel buffer ions continue moving in the stacking gel, but when the glycine ions enters the pH 6.5 of the stacking gel, they become zwitterions with a net charge of zero, and hence stop motion toward the anode. The electrical resistance in the stacking gel then increases since the number of ions moving through the stacking gel decreases. To maintain constant current throughout the circuit, there will be a localized increase in the voltage in the stacking gel (from Ohms Law, V=iR). This will cause the proteins to migrate quickly and all stack in a single, very thin disc right behind the Cl- ions in the stacking gel (which are in front because they have the highest charge density and electrophoretic mobility of any ion in the stacking gel). The proteins will not pass the Cl- ions since if they did, they would immediately slow down since they would no longer be in an area of diminished charged carriers and higher voltage. At the stacking gel/running gel interface, the proteins can not all migrate at the same speed, due to sieving effects of the more concentrated gel, and hence will be separated in the running gel. The glycine eventually enters the running gel, assumes its fully charged state at that pH (8.7), will pass the proteins, and restore the deficiency in charge that occurred in the stacking gel.
Animation of Polyacrylamide Gel Electrophoresis
![]() Java Applet: Protein
Electrophoresis
Java Applet: Protein
Electrophoresis
Detection of proteins in the gel:
Most proteins do not absorb visible wavelengths of light, and hence will not be visible during the course of electrophoresis. To ensure that the proteins are not eluted from the gel into the lower buffer reservoir, a small molecular weight, anionic dye, bromophenol blue is added to the protein before it is placed on the gel. The electrophoresis is halted when the dye reaches near the end of the running gel. The gel assembly is removed from the electrophoresis chamber, the glass plates separated, and the gel washed into a series of solutions with the goal of rendering the banded proteins visible to the eye.
Several techniques are currently used. The most common is to stain the gel with Coomassie Blue, dissolved in a methanol/acetic acid solution. As discovered in lab two, proteins bind Coomassie Blue, with a concomitant spectral shift in the absorbance properties of the bound dye. The methanol and acetic acid in the dye solution also help to "fix" the protein in the gel, and prevent its diffusion into the solution. After the gel is stained, the background stain in the gel is removed with acetic acid/methanol, leaving the blue colored protein bands. Again, a note of caution: some proteins will not be stained with Coomassie blue. Another common staining technique involves silver staining, which involves the reduction of Ag(I) to elemental silver and its deposition by protein in the appropriate reaction solutions, much as in a photographic process. (Remember in the BCA assay, peptide bonds reduce Cu(II) to Cu(I), which is chelated to BCA.) A developer and fixer solution is required. This technique is 10-50 X more sensitive than Coomassie Blue staining. Pre-electrophoresis fluorescent or radioactive modification of the proteins allows even greater sensitivity. After the electrophoresis of a radiolabeled protein, the gel can be dried, and overlayed with X-ray film for periods as long as months, if necessary, to allow sufficient exposure of the film by a low concentration protein. This visualization techniques is called autoradiography.
Figure: SDS PAGE Gel with MW standards
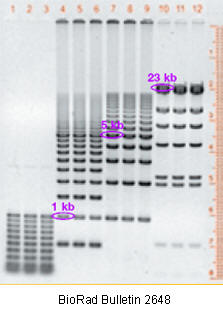
DNA fragments are usually separated on horizontal agarose gel systems, which are much easier to pour.
Figure: DNA fragments separated on an agarose gel
Variations on gel electrophoresis:
Isoelectric focusing: In this technique, a pH gradient is set up within the polyacrylamide gel. This is accomplished by preelectrophoresing a series of low molecular weight molecules containing amino and carboxyl groups called ampholytes. When subjected to an electric field, the most negative of the species will concentrate at the anode, while the most positive will concentrate toward the cathode. The remaining ampholytes will migrate in-between, with the net effect being that the ampholytes migrate to their isoelectric point and set up a linear pH gradient in the gel. A protein applied to the gel will migrate to the pH corresponding to its isoelectric point and stop.
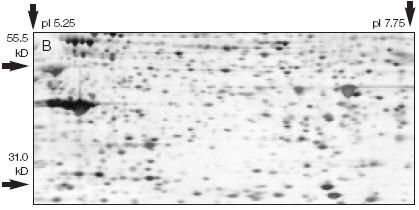
2D electrophoresis: This typically involves subjecting the proteins to isoelectric focusing electrophoresis in a polyacrylamide gel cast in a narrow cylindrical tube. After this electrophoresis, the tube gel is removed, and placed across the top of the stacking gel and subjected to SDS-polyacrylamide gel electrophoresis in a direction 90o from the initial isoelectric focusing experiment. If the proteins were derived from cells labeled with 35Met, representing unique proteins can be obtained from a given cell population.
Figure: 2D Gel
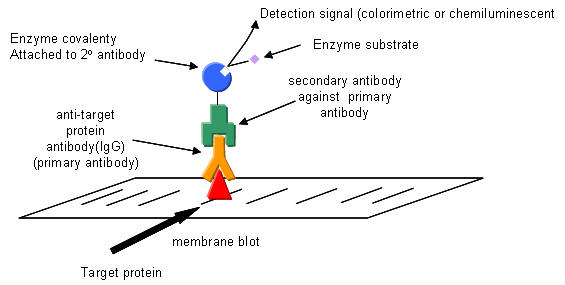
Western blotting: After a standard SDS-slab electrophoresis experiment is run, the gel is overlaid with a piece of nitrocellulose filter paper. The sandwich of gel and filter paper is placed back into an electrophoresis chamber, such that the proteins migrate from the gel into the nitrocellulose, where they irreversibly bind. The filter paper can be removed and soaked in a solution containing a specific antibody to a protein of interest on the nitrocellulose. This protein- antibody complex on the filter paper can then be detected by adding a fluorescently-labeled antibody that binds the first antibody, for instance.
Figure: SDS PAGE gel and Western Blot to detect CKK47
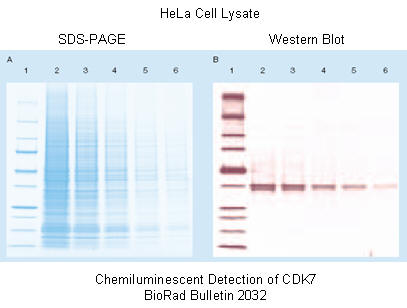
Figure: Detection in Western Blots
Capillary Electrophoresis;
A review of many of the techniques of protein purifications that we have studies in lab.
{kind=link}
{kind=link}
{kind=link}