Determination of Mechanism in Chemistry
Detection of Intermediates
DI3. Trapping Intermediates
Matrix isolation and related methods seek to arrest an intermediate in an otherwise undisturbed state in order to gain direct structural confirmaton about the presence of this species along the reaction pathway. Sometimes, reactive intermediates can't be directly detected because they collapse too quickly into an ensuing structure along the pathway.
In that case, there are sometimes approaches that will allow the intermediate to be trapped as part of another structure. Although the intermediate is perturbed, and would no longer be capable of proceeding to products, the presence of the trapped structure provides compelling evidence that the presumed intermediate did exist at one time.
The presence of radical intermediates has sometimes been inferred because the reaction can be retarded by additives that presumably interfere with radical intermediates. Thiols, for example, readily yield hydrogen atoms because of the relative stability of the sulfur radical. Adding thiols to a reaction suspected of proceeding through a radical mechanism can provide useful confirmation by interfering with the radical intermediate.
![]()
TEMPO (2,2,6,6-piperidine-1-oxyl) is a very effective radical scavenger. Stable enough to be sold commercially, it can be dispensed from a bottle, weighed on a balance, and added to a reaction mixture. It is already a radical so it readily undergoes termination reactions with other radical species. In this case, trapping the intermediate can be accomplished reversibly; the TEMPO-radical adduct can sometimes dissociate homolytically, allowing the original radical species to form again. This approach has been used to gain control over radical polymerizations, slowing the reaction down to prevent random and unwanted termination processes.
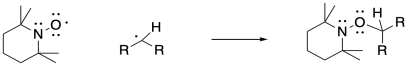
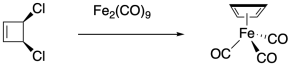
Trapping approaches can be employed to intercept other reactive species. A classic example comes from the quest for cyclobutadiene, the elusive and possibly anti-aromatic four-carbon analog of benzene. Numerous attempts to synthesize this compound failed, giving products that appeared to result from reactive decomposition of the cyclobutadiene (results which themselves implied that cyclobutadiene had formed at some point). Researchers at the University of Texas were able to stabilize cyclobutadiene as an iron(0) complex by generating the reactive molecule in situ.1 This approach reflects a common theme in organometallic chemistry, in which metals are able to stabilize otherwise reactive species.
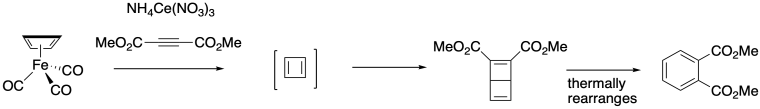
The same laboratory showed that oxidation of the cyclobutadiene complex with cerium(IV) liberated the cyclobutadiene, which could subsequently be trapped by a number of agents via a Diels Alder cycloaddition. The example below was performed using an alkene; the resulting adduct rearranged into a more stable aromatic derivative.
Not only can transition metals stabilize reactive species as ligands, but unstable transition metal species can also be trapped with the appropriate ligand. This approach proved useful in the serendipitously observed C-H activation below; similar reactions with alkyl zirconium amide complexes were discovered in two different labs: one at Berkeley and one at Cornell.3 C-H activation is an important goal for a number of reasons, including applications in organic synthesis and the conversion of methane, typically wasted in petroleum extraction, into useful products.
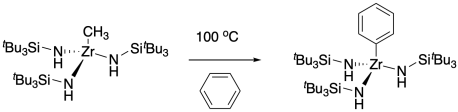
At the time of this discovery, a couple of mechanistic possibilities might have explained the reaction. A process called sigma bond metathesis had been observed several years previously by Patricia Watson at DuPont. In this reaction, the metal-alkyl bond would trade partners with the carbon-hydrogen bond of benzene, going through a rather crowded four-membered ring transition state. Both labs that observed this reaction instead thought a 1,2-elimination of methane was involved, forming a zirconium imide, with C-H activation occuring through addition across the metal-ligand multiple bond.
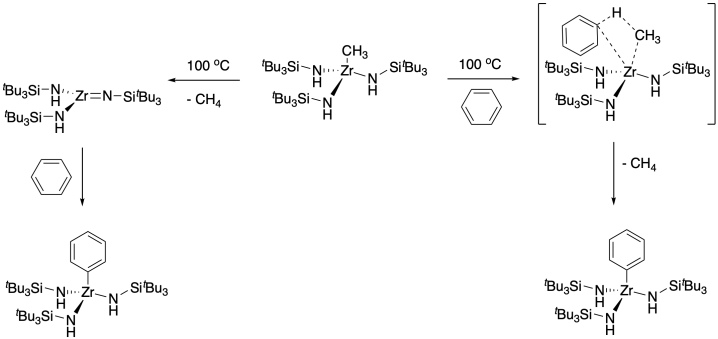
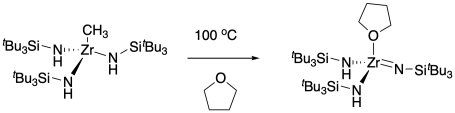
A number of experiments confirmed this latter mechanism, among them a trapping experiment in which the original compound was thermolysed in a donor solvent, THF. The resulting THF adduct of the low-coordinate, highly reactive zirconium imide was structurally confirmed by X-ray crystallography.
Problem DI3.1.
Researchers at Concordia University in Montreal wished to use trapping methods to reveal oxidation of amino acids at very low levels in order to establish protocols for studying oxidative damage in proteins.4 An protected tyrosine was used as a model because it is easily oxidized to give ortho-substituted products. The compound was treated with horseradish peroxidase and TEMPO and the products were analysed by mass spectrometry.
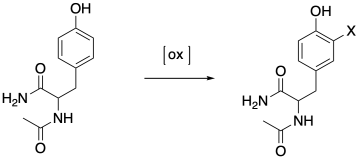
Three different singly protonated products were observed at m/z = 222.8, 378.1 and 443.3. Propose structures for each ion.
Problem DI3.2.
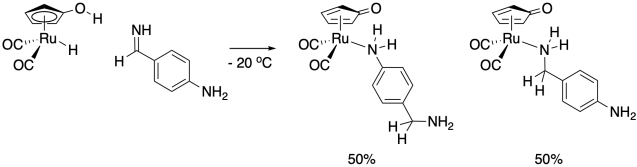
The Casey lab at University of Wisconsin used a trapping experiment to settle a debate about two possible mechanisms for a catalytic hydrogenation mechanism.5 In one mechanism, the aldehyde substrate hydrogen bonds to a ligand hydroxy group, initiating a concerted transfer. In the other mechanism, the cyclopentadiene ligans "slips" to allow aldehyde coordination prior to hydride transfer.
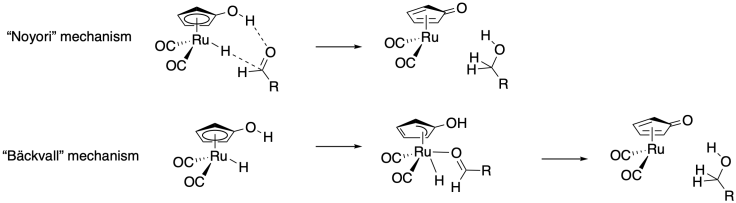
Alcohols don't bind strongly to ruthenium, but amines do, so the Casey lab used an imine substrate in the hopes of trapping the coordinatively unsaturated intermediate. Based on the product ratio, which mechanism seems more likely?
References Cited
1. Emerson, G. F.; Watts, L.; Pettit, R. Cyclobutadiene- and Benzocyclobutadiene-Iron Tricarbonyl Complexes. J. Am. Chem. Soc. 1965, 87, 131-133.
2. Watts, L.; Fitzpatrick, J. D.; Pettit, R. Cyclobutadiene. J. Am. Chem. Soc. 1965, 87, 3253-3254.
3. a) Cummins, C. C.; Baxter, S. M.; Wolczanski, P. T. Methane and Benzene Activation via Transient (tert-Bu3SiNH)2Zr=NSi-tert-Bu3. J. Am. Chem. Soc. 1988, 110, 8731-8733. b) Walsh, P. J.; Hollander, F.; Bergman, R. G. Generation, alkyne cycloaddition, arene carbon-hydrogen activation, nitrogen-hydrogen activation and dative ligand trapping reactions of the first monomeric imidozirconocene (Cp2Zr=NR) complexes. J. Am. Chem. Soc. 1988, 110, 8729-8731.
4. Wright, P. J.; English, A. M. Scavenging with TEMPO To Identify Peptide- and Protein-Based Radicals by Mass Spectrometry: Advantages of Spin Scavenging over Spin Trapping. J. Am. Chem. Soc. 2003, 125, 8655-8665.
5. Casey, C. P.; Bikzhanova, G. A.; Cui, Q.; Guzei, I. A. Reduction of Imines by Hydroxycyclopentadienyl Ruthenium Hydride: Intramolecular Trapping Evidence for Hydride and Proton Transfer Outside the Coordination Sphere of the Metal. J. Am. Chem. Soc. 2005, 127, 14062-14071.
This site is written and maintained by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (with contributions from other authors as noted). It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
Navigation:
Back to Determination of Mechanism
Back to Structure & Reactivity