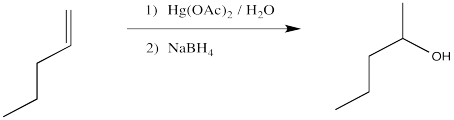
Reactivity in Chemistry
Alkene Addition
EA5.Addition to Coordinated Alkenes
Alkenes can coordinate to transition metals to form alkene complexes. In some cases, coordination of the alkene to a metal leaves it susceptible to reaction with a nucleophile such as water.
The classic case of nucleophilic donation to a coordinated alkene occurs with mercury (II) salts such as mercuric chloride, HgCl2, or mercuric acetate, Hg(OAc)2. The reaction, or rather the sequence of reactions, is called oxymercuration - demercuration or oxymercuration - reduction.
Figure EA5.1. Hydration of an alkene using aqueous Hg2+.
Problem EA5.1.
Compare the product of the reaction above to that obtained from treatment of 1-pentene with aqueous sulfuric acid.
We will break the two different reactions in this sequence apart and focus only on the first one: oxymercuration. This reaction qualifies as an electrophilic addition because, as in the previous cases, it begins with donation of a π-bonding pair to an electrophile. In this case, we will consider the electrophile to be aqueous Hg2+ ion.
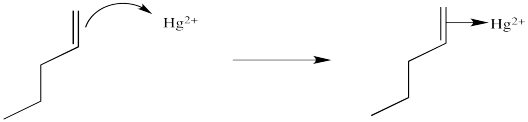
Figure EA5.2. Formation of an alkene complex, shown using dative bond formalism.
That electrophilic addition (from the alkene's perspective) results in the formation of an alkene complex. In reality, the mercury ion is also coordinated by several water molecules, but we will ignore them for simplicity.
You may know that alkene complexes are not observed with d0 transition metals. Although π-to-metal donation is the key event in the formation of such complexes, the alkene is just a little more sticky if the metal has d electrons. These electrons are able to "back-donate" into the alkene portion of the complex, adding extra stability to the interaction.
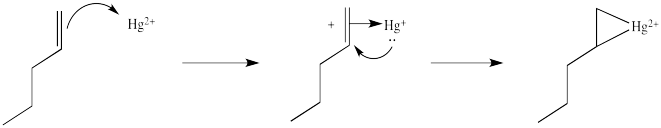
Figure EA5.3. Formation of an alkene complex, shown with back-donation from the metal.
This situation is something like formation of a cyclic bromonium ion. Remember, when an alkene donates to a bromine electrophile, the Br+ can use its plentiful electrons (it has 34 of them) to share with the developing carbocation. That act of sharing stabilizes the carbocation. Instead of forming a regular carbocation, it forms a cyclic bromonium ion, in which the positive charge is distributed over all three atoms in the ring. Hg2+ also has a lot of electrons that it can share with the carbocation (it has 78 of them). Just like with bromine, the carbocation doesn't really form. Instead, we can think of the mercury complex as a three-membered ring, with positive charge distributed over all three atoms in the ring. Organometallic chemists call this kind of ring a "metallacyclopropane", like a cyclopropane that has a metal in it. We can think of it as some superposition or combination of three different resonance structures, each with the charge on a different atom.
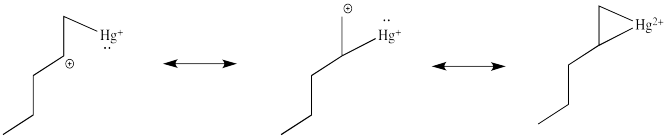
Figure EA5.4. Resonance structures describing charge distribution in a metallacyclopropane.
Notice one of those resonance structure looks particularly bad, because it shows a primary carbocation. You might think that one is a very minor resonance contributor, and you would be right. So we could expect the positive charge to be found mostly on the mercury and on the secondary carbon. In a computational chemistry approach, in which we rely on basic principles of quantum mechanics and let computers churn out high-level calculations to describe the properties of the compound, we would see results showing a little bit of positive charge on the alkene.
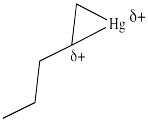
Figure EA5.5. Combining the resonance structures in a metallacyclopropane looks something like this structure.
In the structures above, we have over-emphasized that charge, just to see some of the possibilities in the reaction. When we draw it that way, it looks a lot more like simple addition of electrophile , such as H+, to alkene. We know it's more subtle than that. It's easiest to think of this as a metallacyclopropane, or a cyclic mercurinium ion.
Of course, the next step is donation of a lone pair from a nucleophile to the almost-cationic carbon.
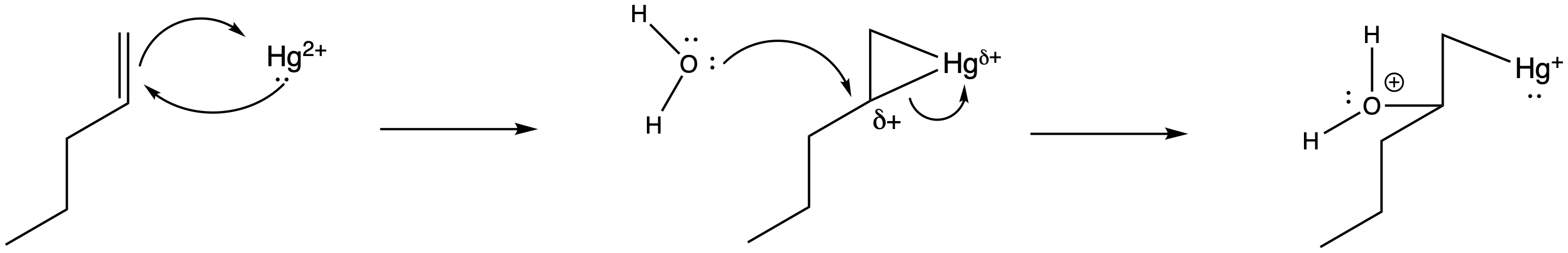
Figure EA5.6. Donation of water to the mercury complexes.
That looks easy. After that, deprotonation would result in the formation of a hydroxy group. The protonated OH group easily loses a proton because it is motivated to lower the positive charge on the electronegative oxygen atom. Any lone pair, such as in a water molecule or acetate group, could remove this proton.
Problem EA5.2.
Suppose deprotonation is carried out by the acetate ion in solution. Draw a mechanism for this step.
How do we know the reaction doesn't happen through this simple cation?
Partly we know that because we know about alkene complexes. There are thousands of examples, structurally characterized by NMR spectroscopy and X-ray crystallography. In addition, we know it isn't a simple cation because nothing like the following scenario plays out during oxymercuration.
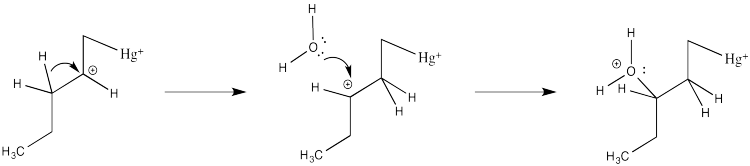
Figure EA5.7. Regular carbocations and hydride shifts are not observed with mercury complexes.
There are no hydride shifts. The cation stays put. The hydroxy group forms right where the alkene used to be. That means there is not a full carbocation like the one shown above.
If there isn't a real carbocation, though, why does the nucleophile end up at one particular end of the alkene? The hydroxy does end up at the position that would form the more stable cation. ( In other words, this reaction results in what is called "Markovnikov addition".)
There are a couple of reasons that could play a role. Foremost, the alkene isn't bound symmetrically. One end is held a little closer to the mercury than the other. Mostly that's because of sterics. Any other ligands on the mercury (such as those water molecules) push that more crowded end away a little bit. That slight asymmetry allows a little more charge to build on the more substitutted end of the alkene, which is therefore more electrophilic.
The final part of the reaction sequence is displacement of mercury from the hydroxyalkylmercury complex, effected through addition of sodium borohydride. The details of the reaction are usually dismissed in textbooks because they have little to do with electrophilic addition, the topic we are focusing on. However, the result is that the mercury is replaced by a hydrogen atom. The metal is converted to silvery, liquid, elemental mercury.
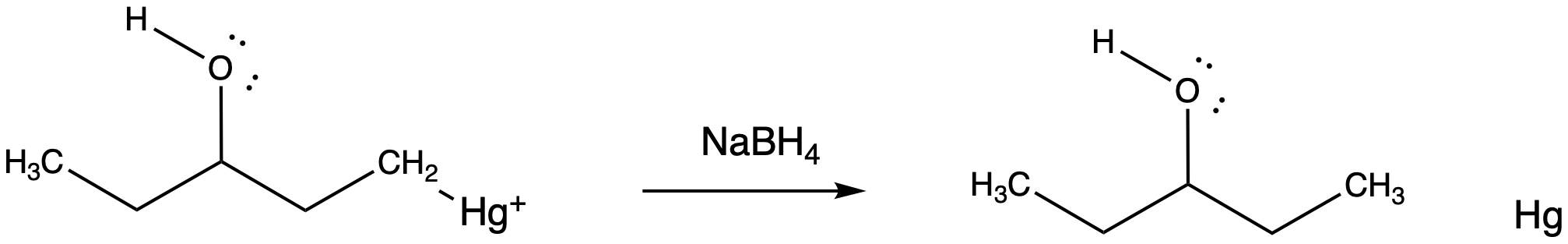
Figure EA5.8. Reduction of the mercury complex.
Problem EA5.3.
Suppose the demercuration reaction takes place via addition of hydride nucleophile to mercury, followed by reductive elimination. Draw this mechanism.
Problem EA5.4.
When mercuration takes place in a ethanol instead of water, an ether product results rather than an alcohol. Work through the mechanism and show the result of mercuration-demercuration in ethanol.
Problem EA5.5.
Show the products of the following reactions.
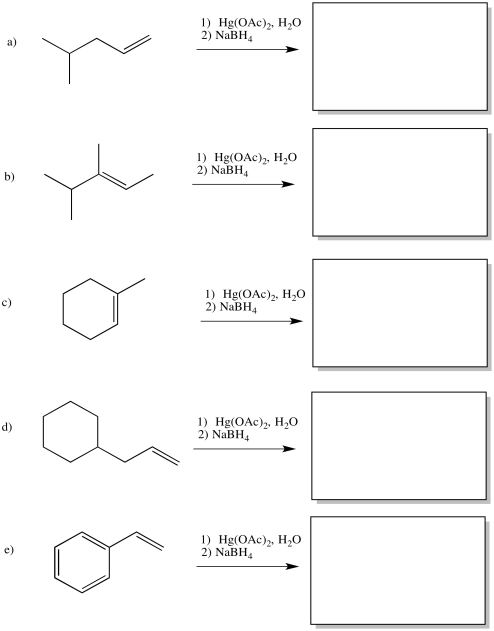
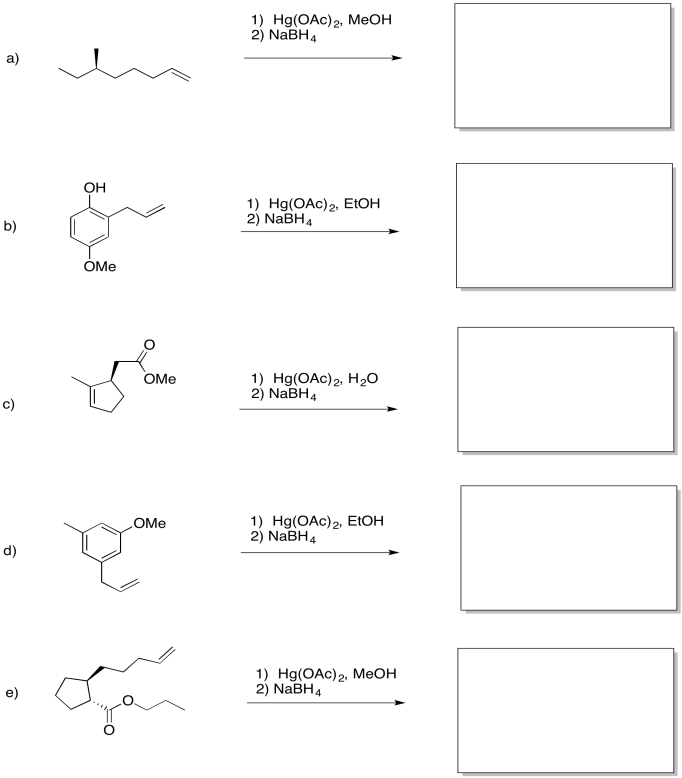
Problem EA5.6.
Predict the products of the following reactions.
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with other authors as noted. It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: