Reactivity in Chemistry
Reduction & Oxidation Reactions
RO12. Organic Redox
The idea of oxidation states is not normally applied to organic compounds, but it can be useful to do so. When we do, we can gain some insight into certain reactions of organic molecules.
For example,carbon dioxide, CO2, can be thought of as having carbon in an oxidized state. If we apply the usual oxidation state rule, carbon dioxide is overall neutral and contains two oxygens, each with 2- charge. To counter that charge, carbon must be in oxidation state 4+.
On the other hand, methane, CH4, can be thought of as having carbon in a reduced state. If we apply the usual oxidation state rule here, methane is overall neutral but contains four protons. That means the carbon must be in a 4- oxidation state. Of course, the carbon does not behave as if it has a minus four charge. But we will see that this sort of exercise can be useful for book-keeping purposes.
Problem RO12.1.
Assign the formal oxidation state to carbon in the following molecules.
a) methanol, CH3OH b) formaldehyde or methanal, CH2O c) carbonate, CO32- d) hydrogen cyanide, HCN
e) ethane, CH3CH3 f) ethene, CH2CH2 g) ethyne, CHCH
The general trend here is that the more bonds there are to oxygen, the more oxidized is carbon. The more bonds there are to hydrogen, the more reduced is carbon.
Oxidation Levels in Organic Compounds
Before we look at redox reactions in organic compounds, we should take a look at a slightly different convention for assessing oxidation states in organic molecules. This convention is the use of oxidation levels rather than oxidation states. An oxidation state refers to an individual atom. An oxidation level refers to a molecule; it reflects the overall sum of the oxidation states of the atoms in that molecule.
Why would we look at things that way? Clearly, we lose some of the detail. Given an oxidation level, you don't immediately know what atom would have what charge. But because these compounds are covalent, rather than ionic, the oxidation state isn't a completely reliable measure of charge, anyway. It is useful for relative comparisons (which carbon would have more buildup of positive charge?) but not absolute measurement (exactly how much charge is on this carbon?). Oxidation level drops that detail but instead gives us a quick comparison between two molecules, such as the reactant and the product of a redox reaction, and that allows us to quickly assess what has happened over the course of the reaction: is this an oxidation, or a reduction?
How does it work? Instead of a focus on atoms, there is a focus on bonds. Look at the number of C-H bonds in a molecule and assign a value of -1 for each bond. Add those numbers into a total. Next, look at the number of C-X bonds in the molecule, in which X is a heteroatom (something other than carbon or hydrogen); that is most often oxygen, but sometimes nitrogen, a halogen, or some other electronegative element. Each of these bonds counts as +1. Add those into the running total. The result is the oxidation level.
For example, look at methanol, CH3OH.
| C-H bonds | 3- |
| C-X bonds | 1+ |
| Oxidation Level | 2- |
Note that we don't worry about O-H bonds. We are just looking at carbon, which is the redox-active atom in organic molecules.
Compare that result to methanal, CH2O.
| C-H bonds | 2- |
| C-X bonds | 2+ |
| Oxidation Level | 0 |
Also compare it to formate ion, HCO2-.
| C-H bonds | 1- |
| C-X bonds | 3+ |
| Oxidation Level | 2+ |
In these cases, the oxidation level is the same as the oxidation state on the carbon atom. That's because there is only one carbon atom in the molecule. If we have additional carbons, oxidation level becomes a much more efficient way of comparing compounds.
Problem RO12.2.
Compare oxidation levels in the following pairs of compounds.
a) CH2CHCH3 and CH3CH2CH3
b) CH3CH2CHO and CH3CH2CH2OH
c) CH3CH(OH)CH2CH2OH and CH3COCH2CHO
Mechanism of Organic Redox
Adding a hydrogen nucleophile to a carbonyl electrophile is routinely referred to as a reduction. For example, adding sodium borohydride to methanal would result in reduction to form methanol. Of course, a hydride is really a proton plus two electrons. We could write an equation for the reduction of methanal that looks a lot like the redox reactions we see in a table of standard reduction potentials.
CH2O + H- + H+ → CH3OH
or
CH2O + 2 e- + 2 H+ → CH3OH
It stands to reason that the opposite reaction, the conversion of methanol to methanal, is a two electron oxidation.
CH3OH → CH2O + 2 e- + 2 H+
We know how to accomplish the reduction of methanal, at least on paper. We just add a complex metal hydride, such as lithium aluminum hydride or sodium borohydride, to the carbonyl compound. After an acidic aqueous workup to remove all the lithium and aluminum compounds, we get methanol. For practical reasons, methanol may be difficult to isolate this way, but that's the general idea of the reaction.
How do we accomplish the reverse reaction: the conversion of an alcohol into a carbonyl?
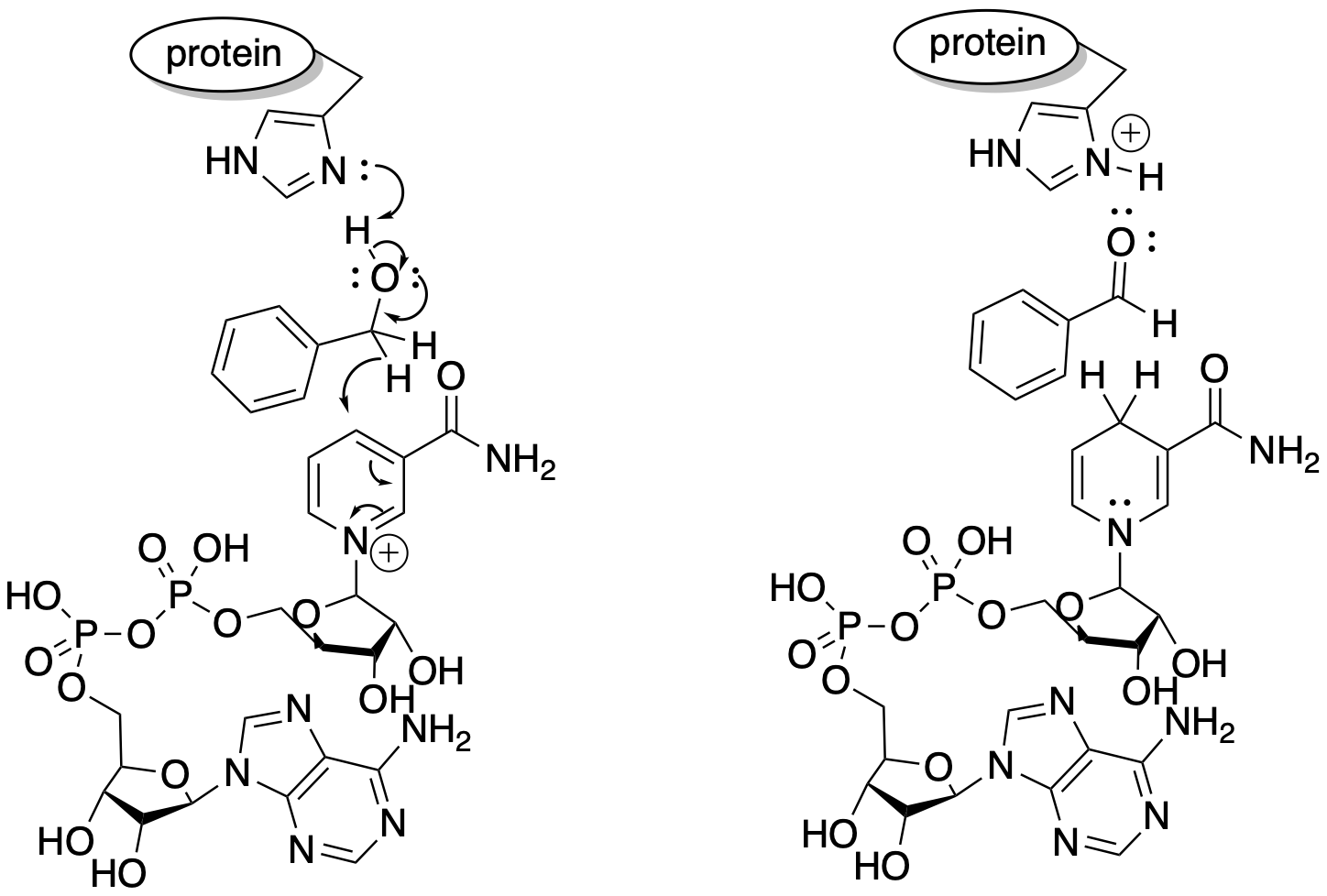
One way would be to provide a hydride acceptor in the reaction, so that we could catch hydride as it comes off the methanol. The most well-known such entity is NAD+, of course. There are biological oxidations that employ NAD+ for this reason.
Figure RO12.1. Oxidation of an alcohol by NAD+.
More generally, the reaction can be accomplished in a number of ways, on paper, by separating out the two tasks involved. We need something to accept the two protons: that's a base. We need something to accept the two electrons: that's an oxidizing agent.
For the latter task, there are a number of high oxidation state transition metal compounds that are quite willing to accept two electrons. One of the most widely employed is Cr(VI), which accepts two electrons to become Cr(IV). A number of other methods are available, having been developed partly to avoid the toxicity of chromium salts, but let's look at the chromium case as an example.
A simple chromium(VI) compound is chromium trioxide. A simple base is pyridine. If we took these two reagents together with benzyl alcohol in a solvent such as dichloromethane, what would happen? OK, you might not want to try this, because chromium trioxide has an alarming capacity to cause spontaneous combustion in organic compounds, but we can do it on paper.
Is chromium trioxide a nucleophile or an electrophile? That Cr(VI) is pretty electrophilic, surely. So what part of the benzyl alcohol is nucleophilic? The oxygen atom. When we mix these things, the oxygen atom would likely coordinate to the chromium.
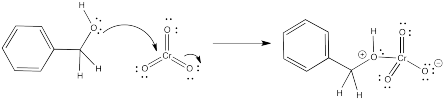
Figure RO12.2. Coordination of an alcohol to CrVI.
When the oxygen atom coordinates to the chromium, the oxygen gets a positive formal charge. It is now motivated to lose a proton. That's what the pyridine is for.
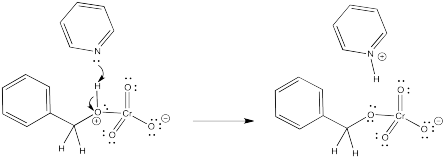
Figure RO12.3. First deprotonation of alcohol coordinated to CrVI.
Now we have accomplished one of the goals of the reaction. We have removed a proton from benzyl alcohol. We have one more proton and two electrons left. The second proton will have to come from the carbon attached to the oxygen; that's the place where we need to form a carbonyl.
But wait a minute. You can't take two protons off the same molecule, can you? And certainly not from two atoms that are right next to each other. Doesn't that generate an unstable dianion?
Not this time. The chromium is there to accept two electrons. We won't generate an anion at all, as far as the benzyl alcohol is concerned. It is oxidized to benzaldehyde.
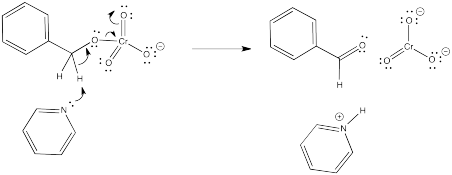
Figure RO12.4. Second deprotonation of alcohol coordinated to CrVI.
Problem RO12.3.
A completely different outcome to this reaction would be obtained in aqueous solution because of the equilibrium that exists between a carbonyl and the geminal diol (or hydrate) in water. Instead of obtaining an aldehyde, a carboxylic acid would be obtained via a second reduction. Provide a mechanism for this reaction.
Oxidation of alcohols is strongly dependent on conditions. In general, there needs to be a hydrogen on the alcoholic carbon (H-C-O-H). If there is no hydrogen on that carbon, the alcohol is pretty difficult to oxidise to a ketone. If there is one hydrogen on that carbon -- that is, if the alcohol is secondary -- then the alcohol becomes a ketone.
If there are two hydrogens on that alcoholic carbon (H2C-OH), i.e. if the alcohol is a primary one, then two different products may result. Removal of just one hydrogen from the alcoholic carbon, and replacement with an additional bond to oxygen, results in formation of an aldehyde. On the other hand, replacement of the second hydrogen from the alcoholic carbon, and replacement with another oxygen, would lead to formation of a carboxylic acid.
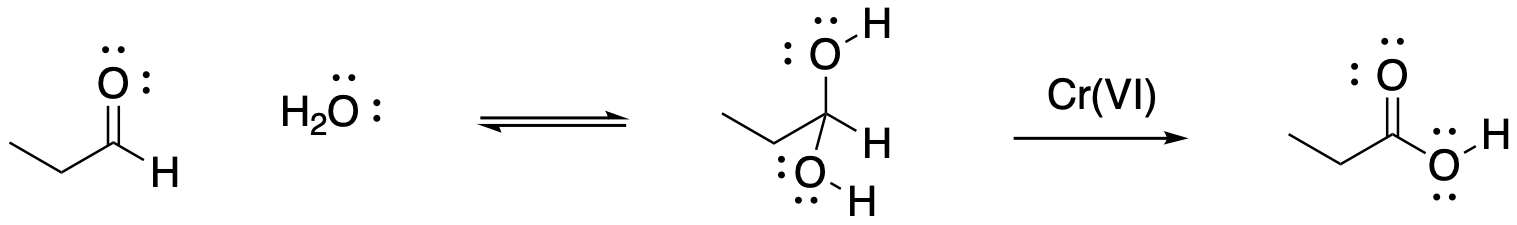
Figure RO12.5. Hydration of an aldehyde leads to a second oxidation.
That second case -- replacement of the second hydrogen with an oxygen -- only happens in aqueous media. The aldehyde that forms after the first oxidation (H-C=O) must become hydrated (H-C(OH)2) in order for the second oxidation to occur, making the carboxylic acid (HO-C=O). As a result, if a primary alcohol is oxidised via a reagent that requires water as a solvent, the carboxylic acid results. If an organic-soluble reagent is used, the reaction stops at the aldehyde.
The most common methods for mildly converting primary alcohols to aldehydes are Swern oxidations and Dess-Martin oxidations. Dess-Martin oxidations employ a high-oxidation state iodine compound -- that's I(V), compared to the more commonly encountered I- ion. The reduction product is an I(III) compound. Swern oxidations employ sulfur in a moderately high oxidation state of zero; the sulfur is reduced to S2- in the Me2S side product.
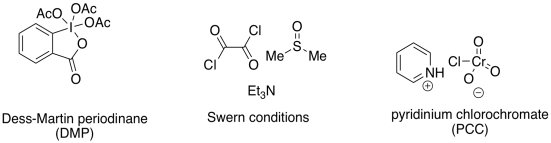
Figure RO12.6. These mild organic oxidants oxidize alcohols but not aldehydes.
In both cases, the oxidation mechanism is similar to the one illustrated with chromium oxide and pyridine. The oxygen donates to the oxidizing atom (the chromium, the sulfur, or the iodine). Deprotonation of the carbon leads to formation of the C=O bond and reduction of the oxidising agent by two electrons.
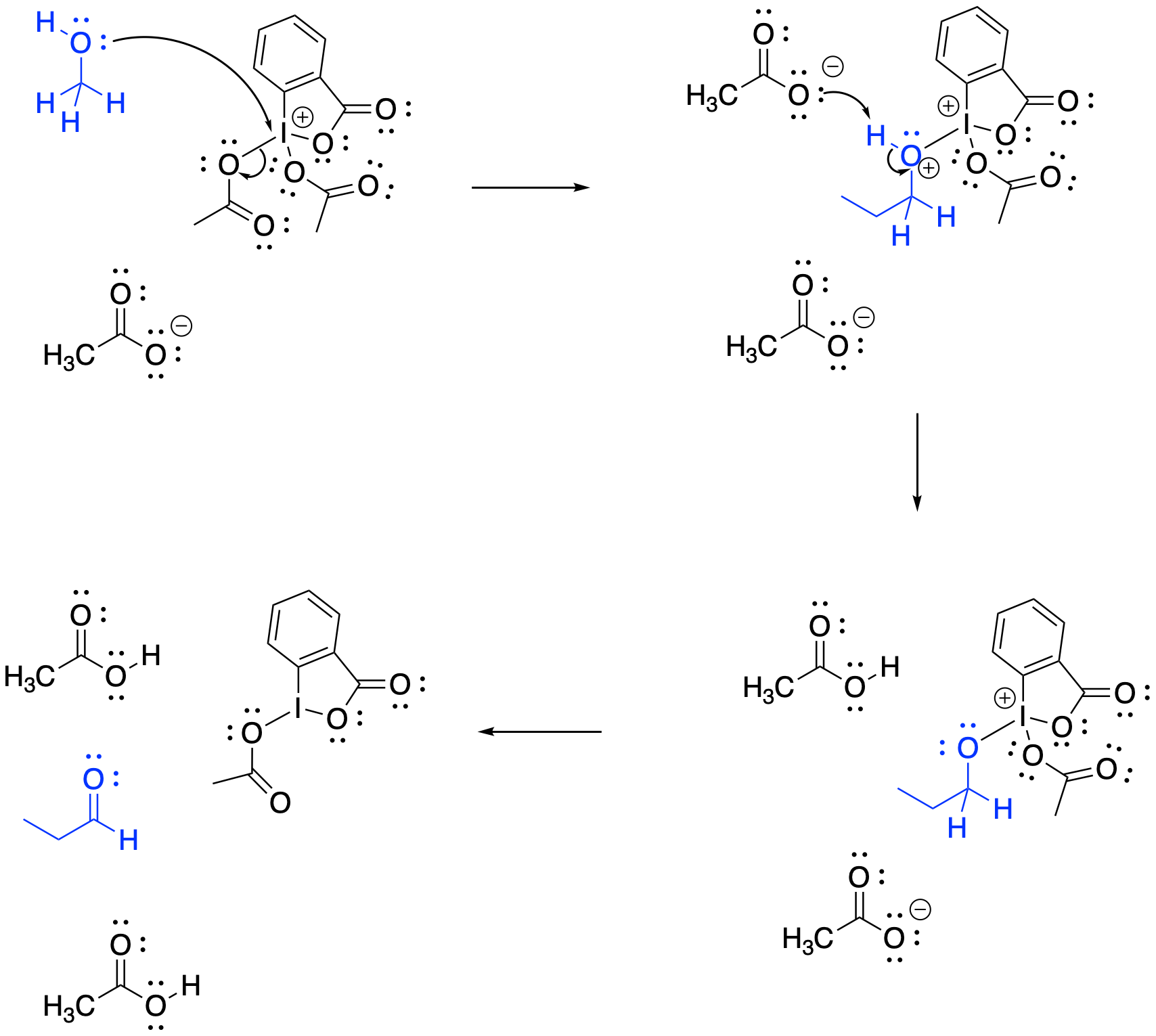
Figure RO12.7. Oxidation of an alcohol by DMP is mechanistically similar to oxidation by chromate.
With Swern oxidations, the mechanism has an added "priming" step, because the thionyl oxidant (S=O) must first be activated; the thionyl oxygen donates to the carbonyl of the oxalyl chloride, (COCl)2.
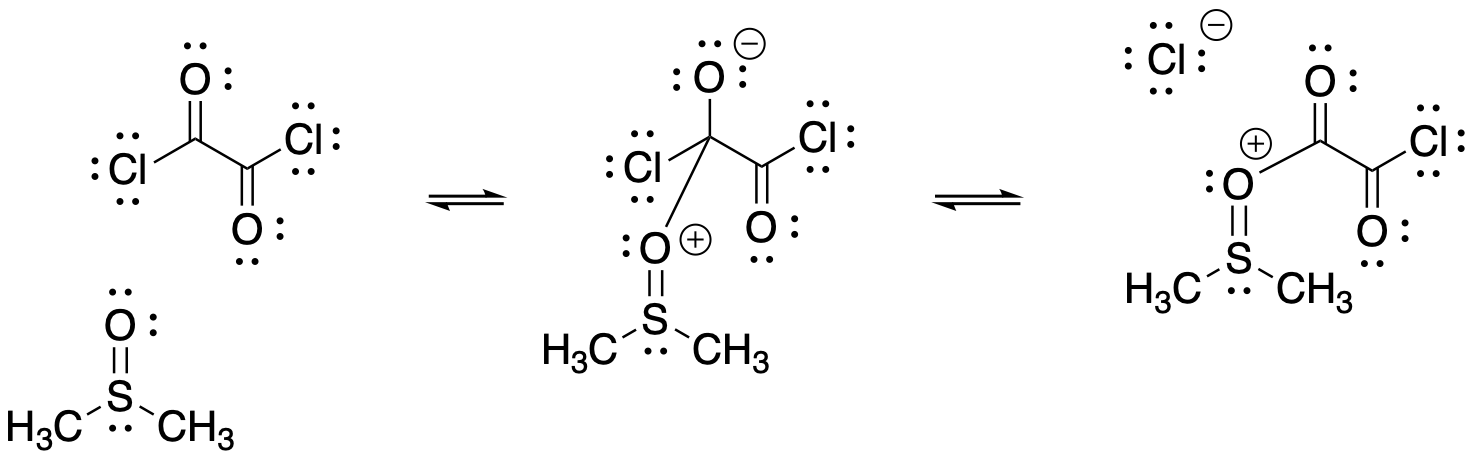
Figure RO12.8. Swern oxidation uses DMSO as an oxidizing agent; it is first pre-mixed to become a better electrophile.
The sulfur is then ready to accept an alcohol donor. Once the alcohol undergoes oxidation, the oxygen from the thionyl group is completley transferred to the oxalyl group, forming both carbon dioxide and carbon monoxide in a subsequent disproportionation reaction.
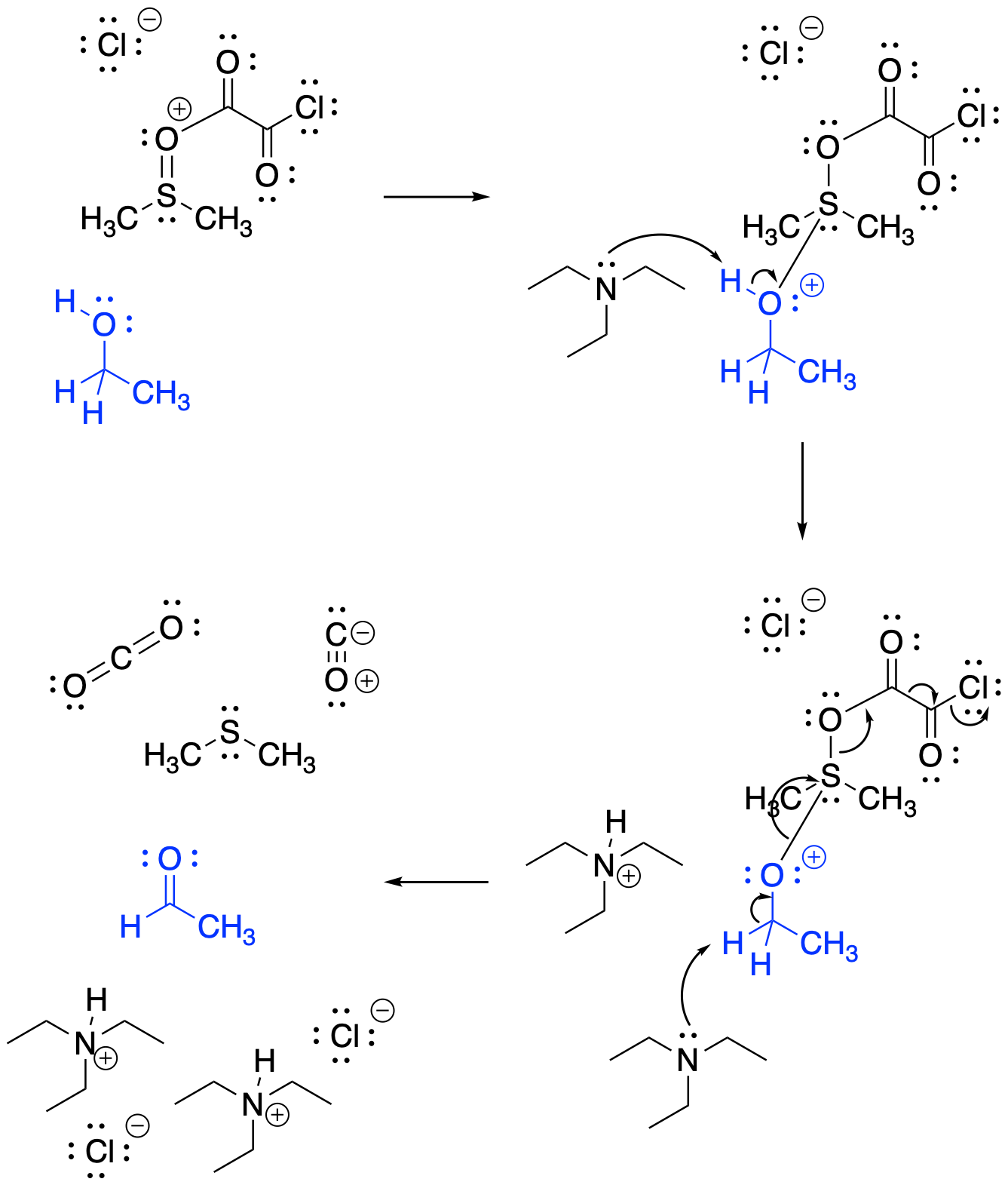
Figure RO12.9. Swern oxidation is driven by decomposition of the oxidizing complex to (CH3)2S, CO2 and CO.
Oddly enough, although PCC looks like a good candidate for aqueous oxidation (it is ionic, after all), it is frequently absorbed onto a solid surface (usually alumina, Al2O3) and used as a heterogeneous catalyst in organic solvent. Like most heterogeneous catalysts, it works rather slowly, but it is pretty selective for aldehyde formation.
Most "ionic" oxidants really are used in the presence of water, and they do convert primary alcohols into carboxylic acids. Examples include sodium chromate, Na2CrO4, dichromate, Na2Cr2O7, and potassium permanganate, KMnO4. These reagents are much harsher than Swern conditions or DMP, however, and they can lead to extensive decomposition of the reactant. Chromates are sometimes prepared as a solution called Jones reagent, in which the oxidant is pre-mixed with sulfuric and and water; the reagent also contains some acetone, to help solubilise the organic compound to be oxidised.
Problem RO12.4.
Fill in the missing products.
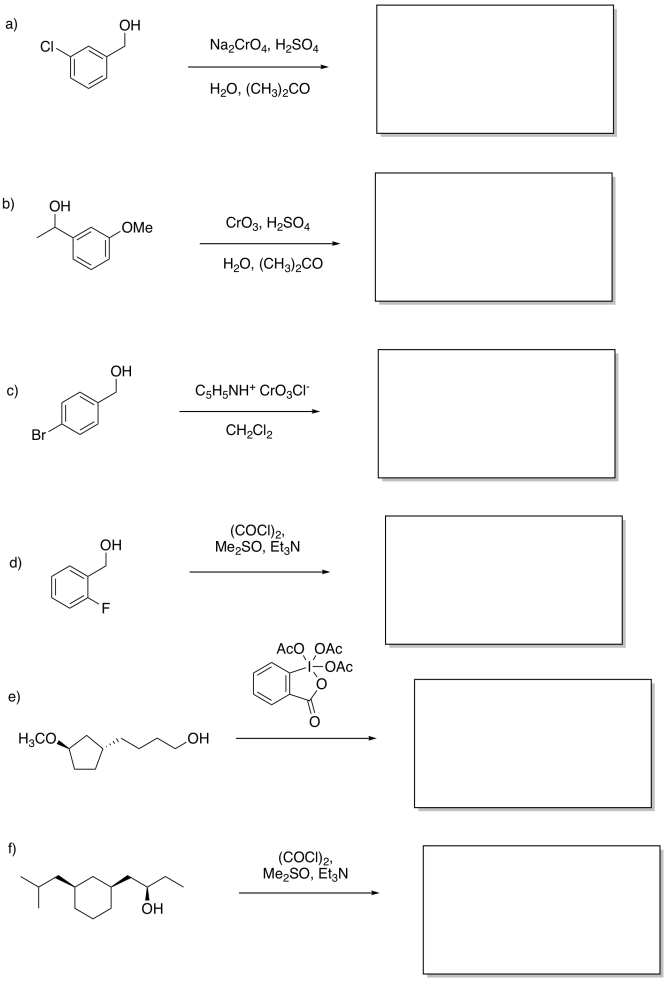
Problem RO12.5.
Fill in the missing reagents.
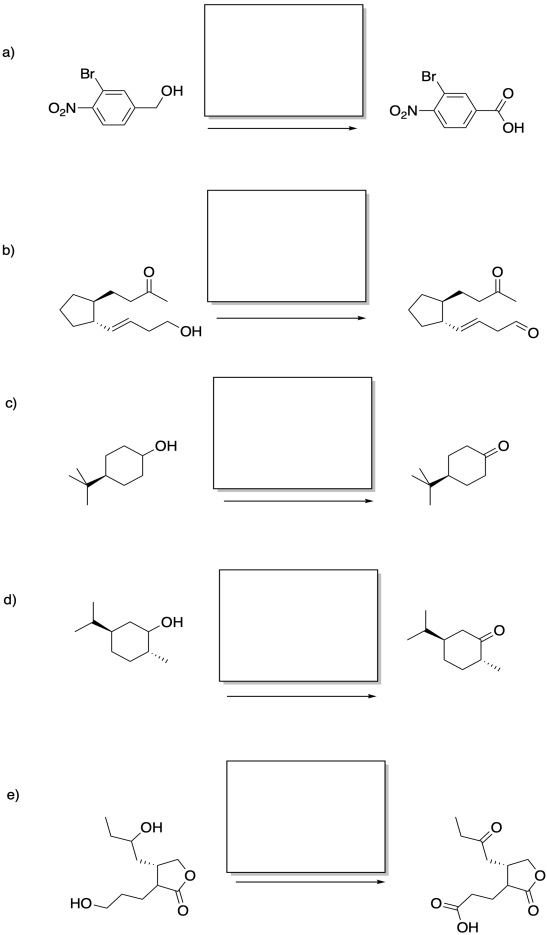
Problem RO12.6.
Fill in the blanks in the following synthesis.
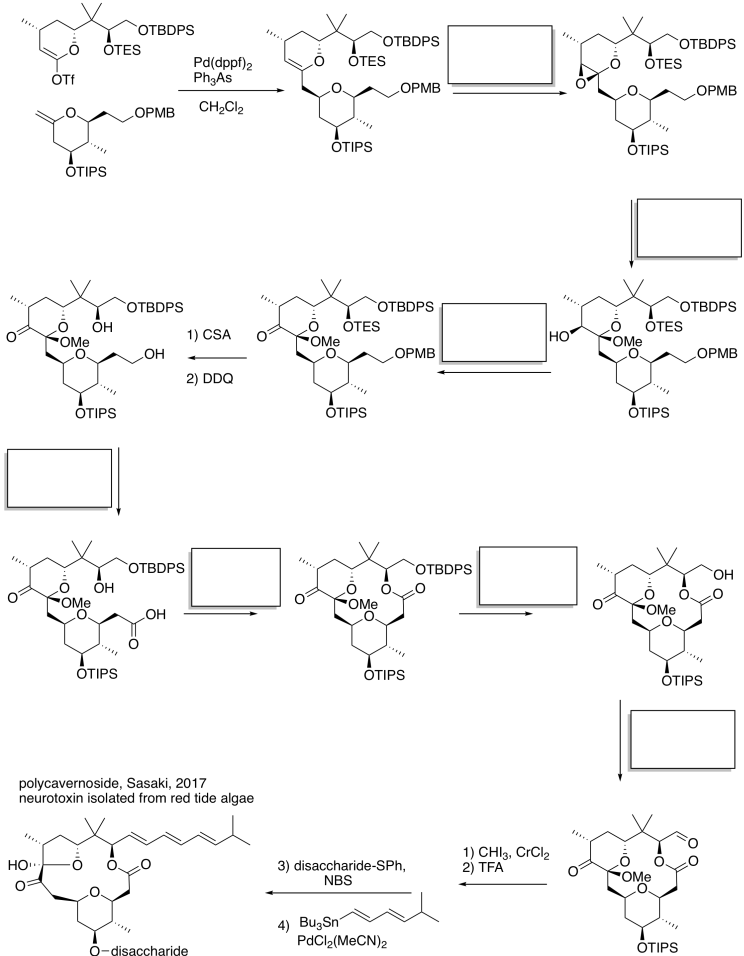
Problem RO12.7.
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with other authors as noted on individual pages. It is freely available for educational use.

Structure & Reactivity in Organic,
Biological and Inorganic Chemistry by
Chris Schaller
is licensed under a
Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: