Biochemistry Online: An Approach Based on Chemical Logic

 Homework
Problems - Literature Learning Module: Binding 1
KEY
Homework
Problems - Literature Learning Module: Binding 1
KEY
Assessment of Biochemistry/Molecular Biology (BMB) Foundational Concepts
Henry Jakubowski, Ph.D., Professor, Chemistry Department, College of Saint Benedict/Saint John's University
Research Paper: Severe Acute Respiratory Syndrome Coronavirus 3C-like Proteinase N Terminus Is Indispensable for Proteolytic Activity but Not for Enzyme Dimerization: Biochemical and Thermodynamic Investigation in Conjunction with Molecular Dynamics Simulations. Shuai Chen, Lili Chen, Jinzhi Tan, Jing Chen, Li Du, Tao Sun, Jianhua Shen, Kaixian Chen, Hualiang Jiang, and Xu Shen. J. Biol. Chem., Vol. 280, Issue 1, 164-173, January 7, 2005
1. Interpret the following CD of the S3 and S3-N proteins. Why did they perform this experiment?
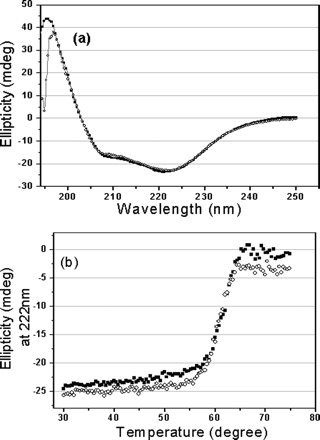
Fig 1. CD spectra of the full-length and N-terminal
deleted SARS 3CLpros. a, far-UV CD spectra of the
full-length (![]() ) and N-terminal
deleted (
) and N-terminal
deleted (![]() ) SARS 3CLpros at
25 �C. b, thermal induced unfolding monitored at 222 nm with a
temperature range from 30 to 75 oC for the full-length (
) SARS 3CLpros at
25 �C. b, thermal induced unfolding monitored at 222 nm with a
temperature range from 30 to 75 oC for the full-length (![]() )
and N-terminal deleted (
)
and N-terminal deleted (![]() ) SARS 3CLpros.
Protein concentrations used in CD experiments were 10 uM,
and all protein samples were prepared in 20 mM sodium
phosphate, pH 7.5, 100 mM NaCl.
) SARS 3CLpros.
Protein concentrations used in CD experiments were 10 uM,
and all protein samples were prepared in 20 mM sodium
phosphate, pH 7.5, 100 mM NaCl.
ANS: Fig. 1 shows the CD spectra of the full-length and N-terminal deleted proteinases. Far-UV spectra of the two proteins give a positive peak at 196 nm characteristic of a β-sheet structure, and dual negative peaks at 209 and 222 nm, respectively, typical of an β-helical structure (Fig. 1a), indicating that the secondary structure of the N-terminal deleted SARS 3CLpro is similar to that of the full-length proteinase, and also similar to those of TGEV and HCoV 229E 3CLpros . This excludes the possibility of structural misfolding caused by N-terminal deletion (residues 1-7). Thermal induced unfolding of the two proteins was performed by monitoring the continuous changes of CD signals at 222 nm within temperatures ranging from 30 to 75 oC, the results are shown in Fig. 1b. As Fig. 1b indicates, the N-terminal deletion does not dramatically affect the denaturation behavior of the proteinase, and the N-terminal deleted proteinase adopts a thermal induced unfolding profile similar to that of the full-length proteinase. The transition temperature (Tm) of the N-terminal deleted proteinase was estimated as 60.5oC, which is very close to that of the full-length proteinase, 61.4oC. All these results imply that N-terminal deletion has not changed the folding manner and thermal stability of the proteinase. They did the experiment to rule out the possibility that the truncated protein folded in a completely different manner.
2. The enzymatic activity of the two proteins was studied using a novel fluorescent substrate (reactant), EDANS-VN_STLQSGLRK(Dabcyl)-M, which is totally hydrolyzed by papain-like protases. This is a short peptide with an amino terminus dansyl fluorophore and a C terminus Dabcyl group which does not fluoresce but can be excited by the excited state dansyl fluorophore ,which can transfer energy to the Dabcyl group in a non-radiative process, but only if it is very close in space to the dansyl group. A graph of enzyme activity is shown below.
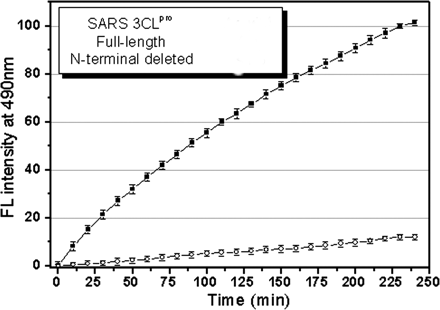
Fig. 2. Representative fluorescence profiles of
hydrolysis of the fluorogenic substrate by SARS 3CLpros. The
fluorogenic substrate at a concentration of 10 uM
was incubated with 1 uM full-length ()
or N-terminal deleted (![]() ) SARS 3CLpro
in 20 mM sodium phosphate, pH 7.5, 100 mM
NaCl, 5 mM DTT, 1 mM EDTA, at 25
o C. Increase of emission fluorescence intensity at 490 nm wavelength was
recorded at 10-min intervals,
) SARS 3CLpro
in 20 mM sodium phosphate, pH 7.5, 100 mM
NaCl, 5 mM DTT, 1 mM EDTA, at 25
o C. Increase of emission fluorescence intensity at 490 nm wavelength was
recorded at 10-min intervals, ![]() EX
= 340 nm. The emission spectrum was recorded for more than 4 h. The initial
reaction velocity (
EX
= 340 nm. The emission spectrum was recorded for more than 4 h. The initial
reaction velocity (![]() 0) was
determined from the linear portion of the progress curve, which corresponded to
between 2 and 10% hydrolysis of the substrate. The activity of the full-length
SARS 3CLpro was taken as 100%.
0) was
determined from the linear portion of the progress curve, which corresponded to
between 2 and 10% hydrolysis of the substrate. The activity of the full-length
SARS 3CLpro was taken as 100%.
a. Give a molecular explanation of why the fluorescence increases on S3 cleavage of the substrate
ANS. When the fluorogenic peptide is cleaved, the fluorophore, dansyl, is separated in space from the dabcyl group, so the energy of the excited state dansyl can not be transferred to the dabcyl group. Hence it loses energy through fluorescence in the absence of this quenching mechanism.
b. What conclusions can you draw concerning the different activity of S3 and S3-N towards the substrate?
ANS: Hydrolysis of the fluorogenic substrate by the full-length SARS 3CLpro results in an appreciable increase in emission fluorescence intensity at the wavelength of 490 nm. The fluorescence profile following hydrolysis of the substrate is depicted in Fig. 2. As a control, the incubation of the substrate in the assay buffer in the absence of 3CLpro shows no fluorescence intensity change over time (data not shown). The result implicates that the fluorogenic substrate could be efficiently hydrolyzed by the full-length SARS 3CLpro. The initial reaction velocity (v0) was determined as 0.14 uM/min from the linear portion of the progress curve.
As expected, the fluorescence increase following hydrolysis of the substrate by the N-terminal deleted SARS 3CLpro is below the detection limit of the activity assay (Fig. 2), suggesting that the N-terminal deleted 3CLpro is almost completely inactive. This result strongly demonstrates the indispensability of the N terminus for enzymatic activity of SARS 3CLpro
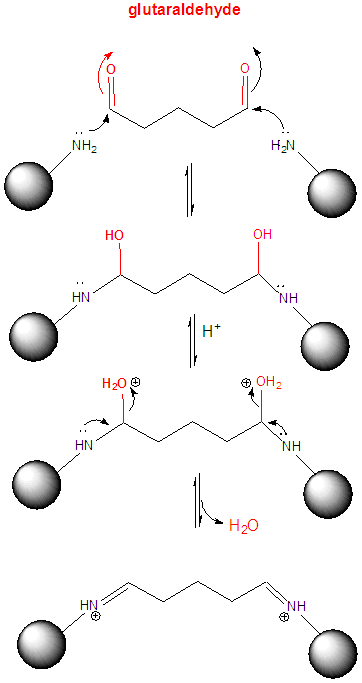
3. One way to determine if a protein can dimerize is to introduce covalent bonds between adjacent proteins subunits in potential dimer or multimer. One such cross-linking reagent is glutaraldehye, which is shown below.

a. Draw a mechanism to show how it could react with Lys residues on two adjacent proteins to crosslink them.
SDS-PAGE gels were run for both S3 and S3-N, and are shown below.
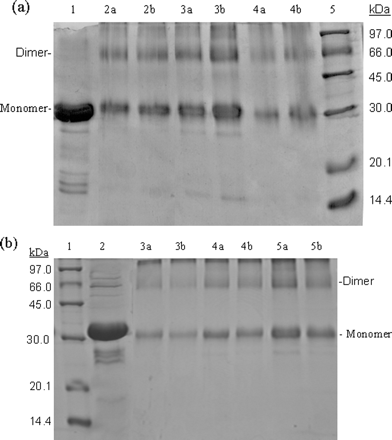
Fig 3. SDS-PAGE (10% acrylamide) profiles of
glutaraldehyde cross-linked SARS 3CLpros.
a, cross-linking
analyses of the full-length SARS 3CLpro. Lane 1, untreated 3CLpro
(7 mg/ml); lane 2a, 3CLpro (2 mg/ml) cross-linked by 0.01%
glutaraldehyde; lane 2b, 3CLpro (2 mg/ml) cross-linked by 0.1%
glutaraldehyde; lanes 3a and 3b, 3CLpro (7 mg/ml)
cross-linked by 0.01 and 0.1% glutaraldehyde, respectively; lanes 4a and
4b, 3CLpro (1 mg/ml), 0.01 and 0.1% glutaraldehyde; lane 5,
molecular weight SDS calibration kit protein standards are as follows:
phosphorylase b (97.0 kDa), albumin (66.0 kDa), ovalbumin (45.0 kDa),
carbonic anhydrase (30.0 kDa), trypsin inhibitor (20.1 kDa),
![]() -lactalbumin (14.4 kDa).
b,
cross-linking analyses of the N-terminal deleted SARS 3CLpro. Lane
1, protein standards; lane 2 untreated 3CLpro (7 mg/ml);
lanes 3a and 3b, 3CLpro (1 mg/ml), 0.01 and 0.1%
glutaraldehyde, respectively; lanes 4a and 4b, 3CLpro
(2 mg/ml), 0.01 and 0.1% glutaraldehyde; lanes 5a and 5b, 3CLpro
(7 mg/ml), 0.01 and 0.1% glutaraldehyde.
-lactalbumin (14.4 kDa).
b,
cross-linking analyses of the N-terminal deleted SARS 3CLpro. Lane
1, protein standards; lane 2 untreated 3CLpro (7 mg/ml);
lanes 3a and 3b, 3CLpro (1 mg/ml), 0.01 and 0.1%
glutaraldehyde, respectively; lanes 4a and 4b, 3CLpro
(2 mg/ml), 0.01 and 0.1% glutaraldehyde; lanes 5a and 5b, 3CLpro
(7 mg/ml), 0.01 and 0.1% glutaraldehyde.
b. What conclusions can you draw concerning the ability for the two proteins to form dimers??
ANS: When incubated with 0.01% glutaraldehyde, the full-length proteinase at a concentration of 1 mg/ml displayed a form of monomer with one other species around 66.0 kDa (Fig. 3a, lane 4a), corresponding to the dimer of the proteinase. At the higher concentration of glutaraldehyde (0.1%), the full-length SARS 3CLpro (1 mg/ml) showed a similar cross-linking pattern (Fig. 3a, lane 4b), indicating that the amount of glutaraldehyde was sufficient throughout and no artificial cross-linking occurred. With protein concentration increasing, the dimeric form increased and was almost equivalent to the amount of the monomeric form at higher concentration (7 mg/ml) (Fig. 3a, lanes 2 and 3). These results suggest that dimerization of the full-length SARS 3CLpro is concentration-dependent.
Most strikingly, similar results were obtained with the N-terminal deleted proteinase. The dimeric form of the N-terminal deleted SARS 3CLpro also existed within a wide range of protein concentrations. When incubated with 0.01 or 0.1% glutaraldehyde, the N-terminal deleted 3CLpro at a concentration of 1 mg/ml also displayed forms that migrated as monomer and dimer (Fig. 3b, lanes 3a and 3b), whereas the monomer was the predominant species at a relatively low protein concentration. When the protein concentration increased to 2 mg/ml, the amount of the dimeric species was fortified (Fig. 3b, lanes 4a and 4b). At higher concentration (7 mg/ml), the band corresponding to the monomer of the N-terminal deleted 3CLpro was only slightly thicker than that of the dimer (Fig. 3b, lanes 5a and 5b). These results indicate that the N-terminal deletion seems to have no impact on the concentration-dependent dimerization of 3CLpro. Obviously, this finding is not fully consistent with the conclusion reported previously, which supposed that the N terminus plays an important role in both dimerization and formation of the active site of SARS 3CLpro
4. To further study the solution behavior of the protein, they were subjected to gel filtration (also called size exclusion chromatgraphy) on Superdex 75. Results are shown in Figure 4 below.
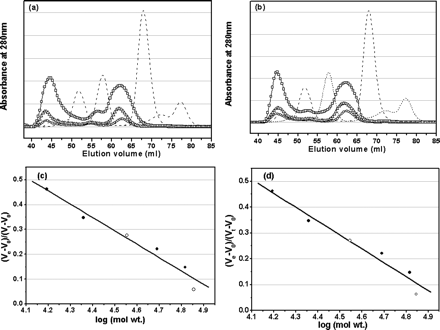
Fig 4. Analyses of monomer-dimer equilibria of SARS 3CLpros
by SEC. a, elution profiles of the full-length SARS 3CLpro
at neutral pH (7.5) and concentrations of 1 (![]() ),
2 mg/ml (
),
2 mg/ml (![]() ), and 7 mg/ml (
), and 7 mg/ml (![]() ).
Elution profiles of four marker proteins, ribonuclease A (15.6 kDa),
chymotrypsinogen A (22.8 kDa), ovalbumin (48.9 kDa), and albumin (65.4 kDa), are
also shown as dashed lines. The dimer/monomer ratios for the
concentrations of 1, 2, and 7 mg/ml are 0.68, 0.71, and 0.84, respectively.
b,
elution profiles of the N-terminal deleted SARS 3CLpro at neutral pH
(7.5) and concentrations of 1 (
).
Elution profiles of four marker proteins, ribonuclease A (15.6 kDa),
chymotrypsinogen A (22.8 kDa), ovalbumin (48.9 kDa), and albumin (65.4 kDa), are
also shown as dashed lines. The dimer/monomer ratios for the
concentrations of 1, 2, and 7 mg/ml are 0.68, 0.71, and 0.84, respectively.
b,
elution profiles of the N-terminal deleted SARS 3CLpro at neutral pH
(7.5) and concentrations of 1 (![]() ), 2 (
), 2 (![]() ),
and 7 mg/ml (
),
and 7 mg/ml (![]() ). Elution profiles of
marker proteins are shown as dashed lines. The dimer/monomer ratios for
the concentrations of 1, 2, and 7 mg/ml are 0.65, 0.66, and 0.80, respectively.
In the SEC-FPLC experiment, each protein sample was loaded to a HiLoad 16/60
Superdex 75 prep grade column pre-equilibrated with the buffer and then eluted
at a flow rate of 1 ml/min with a detection of absorbance at 280 nm. The buffer
used was 20 mM sodium phosphate, pH 7.5, 100 mM
NaCl, 5 mM DTT, 1 mM EDTA.
Standard calibration curves were used to determine the molecular masses of
monomeric and dimeric form of the full-length (c) and N-terminal deleted
(d) SARS 3CLpros. Elution volumes of the full-length and
N-terminal deleted proteinases are indicated by open circles (
). Elution profiles of
marker proteins are shown as dashed lines. The dimer/monomer ratios for
the concentrations of 1, 2, and 7 mg/ml are 0.65, 0.66, and 0.80, respectively.
In the SEC-FPLC experiment, each protein sample was loaded to a HiLoad 16/60
Superdex 75 prep grade column pre-equilibrated with the buffer and then eluted
at a flow rate of 1 ml/min with a detection of absorbance at 280 nm. The buffer
used was 20 mM sodium phosphate, pH 7.5, 100 mM
NaCl, 5 mM DTT, 1 mM EDTA.
Standard calibration curves were used to determine the molecular masses of
monomeric and dimeric form of the full-length (c) and N-terminal deleted
(d) SARS 3CLpros. Elution volumes of the full-length and
N-terminal deleted proteinases are indicated by open circles (![]() )
and for marker proteins by filled circles. Ve and
V0 are peak elution volume and column void volume,
respectively, and Vt is the total bed volume.
)
and for marker proteins by filled circles. Ve and
V0 are peak elution volume and column void volume,
respectively, and Vt is the total bed volume.
a. Why did they vary the concentration of each protein.
ANS: To see if the dimerization was concentration dependent, which would be expected if there was a simple equilibrium between monomer and dimer (with no higher polymers), characterized by a specific Kd.
b. What conclusions can you draw concerning the ability for the two proteins to form dimers?
In order to gain more insight into the dimer-monomer equilibrium of SARS 3CLpro in solution and to exactly evaluate the role of the N-terminal deletion in 3CLpro dimerization, SEC analyses on a HiLoad 16/60 Superdex 75 prep grade column were carried out. The result is represented in Fig. 4. The column was first calibrated with four marker proteins for evaluation of the elution profiles of the protein samples. In the gel filtration experiments under conditions similar to those for chemical cross-linking SDS-PAGE data collection, the physical states corresponding to native dimeric and monomeric 3CLpros were observed within a wide range of protein concentration. For the full-length 3CLpro (1 mg/ml) at neutral pH (7.5), two peaks centered at 44.57 and 62.14 ml were observed (Fig. 4a). The fact that the peak at 62.14 ml is between the retention volumes of 57.22 and 67.89 ml for the two molecular mass marker proteins of 48.9 and 22.8 kDa under similar conditions suggests that this peak corresponds to the monomer state of SARS 3CLpro (35.8 kDa). The other peak centered at 44.57 ml is lower than the retention volume of 51.72 ml for the molecular mass marker protein of 65.4 kDa, which is well attributed to the dimeric species of 3CLpro. This observation thus indicates that the full-length 3CLpros under the conditions studied have both forms of monomer and dimer. The amount of the dimer and monomer forms could be quantified by the integrated area value of two corresponding peaks.
c. The integrated areas of the peaks give dimer/monomer ratios of 0.68, 0.71 and 0.84 for protein concentrations of 1, 2, and 7 mg/ml. Write the chemical and mathematical equilibrium expression for the process and using these, explain if the variation of the dimer/monomer ratio is expected.
Monomer (M) + Monomer (M) <=> Dimer (D)
KD = [M][M]/[D]
This equilibrium is different that the simple M + L <=> ML one that we have encountered before. In that case, we held total M constant, varied L, experimentally determined ML, and then used many methods to extract the KD, including fitting the ML vs L (or Y vs L) data to a hyperbolic nonlinear fit, a double reciprocal plot, or a Scatchard plot. In this case, both M and L, (which in this case is M also) are varying. Nevertheless, you would still expect a graph of D vs M to show basic saturation but since you are constantly adding M, free M will always be available, so the saturation part of the curve will not have a slope a zero but some small positive number as shown below.
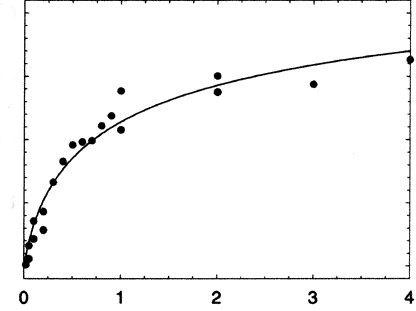
There are several equations that can be used to describe the curve. Here is one:
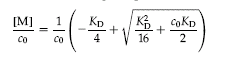 J.
Mol. Biol. (2003)
334,
403�419
J.
Mol. Biol. (2003)
334,
403�419
where co = the total concentration of protein, and [M] is the concentration of free monomoer.
Yes, based on the equations above, which would clearly show It again that 3CLpro dimerization is concentration-dependent. This is in agreement with the results obtained from glutaraldehyde cross-linking SDS-PAGE determinations.
5. The monomer/dimer interaction was then studied by isothermal titration calorimetry. In the experiments, concentrated protein (around 500 - 600 μM or 16-17 mg/ml) was injected into a buffer solution and enthalpy changes measured. Results are shown below. Note that all injections of the protein give + enthalpy values indicated the process is endothermic.
5. 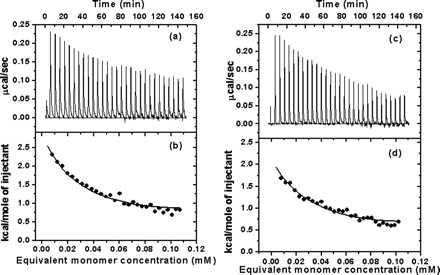
Fig 5. Typical calorimetric dilution data measured by ITC. a, the calorimetric dilution profile for full-length SARS 3CLpro dimers. Consecutive 10 �l volumes of 572.5 �M 3CLpro (approx 17 mg/ml) were diluted into 1.43 ml of 20 mM sodium phosphate, pH 7.5, 100 mM NaCl, 1 mM EDTA, at 25 oC (except an initial injectant of a 3-�l volume). Note that the protein was dissolved in the same buffer solution. b, integrated dilution heat effects of full-length SARS 3CLpro, with theoretical fits by a simple monomer-dimer model to yield a Kd of uM and a dimerization enthalpy change (ΔHodimer) of -8.283 + 0.103 kcal/mol. c, the calorimetric dilution profile for N-terminal deleted SARS 3CLpro. Endothermic responses for sequential 10 ul injections of 544.6 uM 3CLpro into buffer were measured under the same conditions, except a 3-ul initial injection. d, integrated dilution heat effects of N-terminal deleted SARS 3CLpro , with theoretical fits to yield a Kd of 262 + 15 uM and a ΔHodimerization of -6.893 + 0.25 kcal/mol.
a. Write a chemical equation that clearly shows what process occurs when the first injection of protein S3 is made into the calorimeter cell. Calculate the initial concentration of S3 at the instant (t=0) it is injected into the calorimeter cell. What is the state of the diluted protein at t=0 and at the end of the time increment indicated by return of the enthalpy curve to the base line value AFTER THE FIRST INJECTION (i.e. at the end of the spike in the enthalpy for the first 10 ul protein injection).?
ANS: They injected 3 ul of the concentrated protein initially, which since it is so concentrated it would exists predominantly in the dimeric form. When injected into 1.42 ml of buffer solution, the protein was diluted 474 fold, making the protein concentration in the cell after the first injection of 572.5 uM/474 = 1.21 uM protein . If the first point in the graph was when 10 uL was injected, the dilution would be 1.43/0.01 = 1 to 143 and the protein concentration in the cell would be 572.5/143 = 4.0 uM protein = 0.004 mM, which looks like the concentration shown for the first injection on the graph. If the Kd is 227 uM, either of these 2 protein concentrations would be much much less than the Kd. At the instant it was injected, t = 0, the protein would be dimeric. It would immediatley dissociate however, given the total protein concentration in the cell, with the associated enthalpy change.
Hence, the only process that would occur on the initial dilution would be dissociation of the dimer, or
D (Dimer) --> 2 M (Monomer)
b. Why does the enthalpy change becomes smaller and smaller (as shown in Fig 5b) as more concentrated protein is added to the calorimetry cells.
ANS: As more is injected, and more of the protein in solution is in the monomeric state, the equilibrium on subsequent injects gets shifted back to the formation of dimer (as the monomer concentration exceeds Kd). Eventualy the dimeric form would no dissociate and no enthalpy change would result on dilution. The effective concentration of monomer would rise. In the graph the enthalpy change occurs when dimers dissociate. The great signal occurs initially. As more and more 10 ul aliquots are added, the monomer builds up and less dimer dissociates
c. Calculate ΔG0 and ΔS0 for the dimerization equilibrium for S3.
ANS: If you know Kd and ΔH0, you can determine the above thermodynamic parameters, since,
ΔG0 = -RTlnKeq = +RTlnKd = ΔH0 -T ΔS0 .
The paper stated that Kd = 227 mM and ΔH0 = -8.28 kcal/mol. :
ΔG0 = -5.0 kcal/mol ; ΔS0 = -11 cal/mol deg K
d. What conclusions can you draw concerning the ability for the two proteins to form dimers?
ANS: The typical calorimetric dilution profile of the full-length 3CLpro is shown in Fig. 5a, which indicates that the dilution process is endothermic. This is in agreement with the phenomenon derived from the chemical cross-linking SDS-PAGE and SEC analyses that 3CLpro dimerization is concentration-dependent (Figs. 3 and 4). The sequence of dilution injections produces a series of endothermic heat pulses which, after integration and correction for buffer mixing control experiments, gives the absolute heat uptake per injection. In such a dilution series, successive injections become progressively less endothermic as the 3CLpro concentration increases in the sample cell. The resulting thermal dilution profile is consistent with a simple dimer-monomer dissociation model. In a similar way, the calorimetric dilution profile of the N-terminal deleted 3CLpro was obtained, which is shown in Fig. 5, c and d, indicating that dimerization of the N-terminal deleted proteinase is also endothermic and concentration-dependent. Here are the actual tabulated values:
TABLE I
Thermodynamic parameters for SARS 3CLpro dimerization at 25 oC and pH 7.5
|
|||||||||||||||||||||||||
6. Molecular dynamics simulations were made from the pdb file for S3 (in which the unit cell contained a dimer) and models based on it for S3-N. Modeling shows that S3-N forms a dimer but with a reduced substrate binding pocket. To confirm this, a small peptide substrate, TSAVLQ, was synthesized. This represented the N terminal sequence of the best reported peptide substrate for the enzyme. The binding affinity of the peptide for S3 and S3-N was determine by surface plasmon resonance biosensors. S3 or S3-N were immobilized to cells for sensor chip from Biacore. Buffer was continuously flowed over the derivitized chip. The peptide was diluted into the buffer solution and injected in increasing concentrations from 20-500 μM. Binding responses were recorded continuously in resonance units (RU) as sensorgrams. The results are shown below.
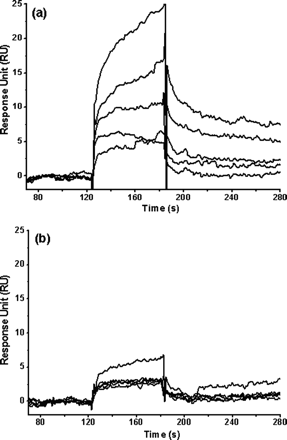
Fig 7. Binding affinity of a 6-amino acid peptide (TSAVLQ) to the full-length and N-terminal deleted SARS 3CLpros determined by SPR. Real time binding affinity measurements of the peptide to the full-length SARS 3CLpro (a) and the N-terminal deleted proteinase (b) using Biacore 3000. Representative sensorgrams were obtained from injections of the peptide at concentrations of 500, 250, 100, 50, and 20 �M (curves from top to bottom). The peptides were injected for 60s (from 120-180 seconds), and dissociation was monitored for more than 120s (from 180 to 300 seconds).
a. Write the chemical equilibrium expression for the protein and peptide interaction showing appropriate rate constants. What constant (Kd, kforward, or kreverse can be determined from data in the range 120-180s? 180-300s?
ANS: In the range 120-180 kforward (or kon) can be determined, while after 180 s, when no more peptide is added, kreverse (or koff) is measured
b. From kforward and kreverse, how could you determine Kd?
ANS: The forward reaction is given by M + L --> ML, and the velocity equation is: vf = kforward[M][L].
The reverse reaction is given by ML --> M + L, and the velocity equation is: vr = kreverse[ML] .
At equilbrium, vf = kforward[M][L] = vr = kreverse[ML] . Hence
kreverse/kforward = [M][L]/[ML] = Kd.
c. Compare and interpret the two graphs. Are the data consistent with the other experiments?
For the full-length proteinase (Fig. 7a), the peptide resulted in a significant and dose-dependent increase in SPR response units and presented characteristic fast binding and slow dissociation curves, indicating a rapidly formed and relatively stable proteinase-peptide complex. The peptide concentration series were fitted to a steady-state binding model encoded in the Biacore 3000 evaluation software for binding affinity determination. The dissociation constant (KD value) between the substrate peptide and the full-length proteinase was determined as about 152 �M. For the N-terminal deleted proteinase, a negligible and concentration-independent RU increase was obtained (Fig. 7b), indicating that almost no binding exists between the substrate peptide and the N-terminal deleted proteinase. This result is consistent with the results of the above MD simulation..
 Navigation
Navigation
Return to Biochemistry Online Table of Contents

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.