Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 4 - DNA and the Central Dogma of Biology
BIOCHEMISTRY - DR. JAKUBOWSKI
3/23/16
C1. The Language of DNA
Introduction
In this short chapter you will learn how modern molecular biologists manipulate DNA, the blueprint for all of life. The four letter alphabet (A, G, C, and T) that makes up DNA represents a language that when transcribed and translated leads to the myriad of proteins that make us who we are as a species and as individuals. Let's continue with the metaphor that DNA is a language. To master that language, as with any other language, we need to be able to read, write, copy, and edit that language. If you were using a word processor to find one line in a hundred page document, or one article from one book out of the Library of Congress, you would also need a way to search the large print base available. You might want to compare two different copies of files to see if they differ from each other. From the lab and this online discussion and problem set, you will learn how modern scientists read, write, copy, edit, search, and compare the language of the genome. These abilities, acquired over the last twenty years, have revolutionized our understanding of life and have given us the potential to alter, for good or evil, life itself.
DNA in human chromosomes exists as one long double stranded molecule. It is too long to physically study and manipulate in the lab. Using a battery of enzymes, the DNA of chromosomes can be chemically cleaved into smaller fragments which are more readily manipulable. (Similar techniques are used to sequence proteins, which require overlapping polypeptide fragments to be made.) After the fragments have been made, they must be separated from each other in order to study them. DNA fragments can be separated on the basis of some structural feature that differentiates the fragments from each other. Polarity can not be used since all DNA fragments have negatively charged phosphates in the sugar - phosphate backbone of the molecule. Although each fragment would have a unique sequence, it would be hard to separate all the different fragments, by, for instance, attaching some molecule that binds to a unique sequence in the major groove of a given fragment to a big bead and using that bead to separate out that one unique fragment. You would need a different bead for each unique fragment! The best way to separate the fragments from each other is to base the separation on the actual size of the fragment by using electrophoresis on an agarose or polyacrylamide gel.
A carbohydrate extract called agarose is made from algae. Water is added to the extract, which is then heated. The carbohydrate extract dissolves in the water to form a viscous solution. The agarose solution is poured into a mold (like warm jello) and is allowed to solidify. A plastic comb with wide teeth was placed in the agarose when it was still liquid. When the agarose is solid, the comb can be removed, leaving in its place little wells. A solution of DNA fragments can be placed in the wells. The agarose slab with sample is covered with a buffer solution and electrodes placed at each end of the slab. The negative electrode is placed near the well end of the agarose slab while the positive electrode is placed at the other end. If a voltage is applied across the agarose slab, the negatively charged DNA fragments will move through the agarose gel toward the positive electrode. This migration of charged molecules in solution toward an oppositely charged electrode is called electrophoresis. Pretend you are one of the fragments. To you the gel looks like a tangle cobweb. You sneak your way through the openings in the web as you move straight forward to the positive electrode. The larger the fragment, the slower you move because it is hard to get through the tangled web. Conversely, the shorter the fragment, the faster you move. Using this technique and its many modifications, oligonucleotides differing by just one nucleotides can be separated from each other. In electrophoresis of DNA fragments, a fluorescent, uncharged dye, ethidium bromide, is added to the buffer solution. This dye literally intercalates in-between the base pairs of DNA, which imparts a fluorescent yellow-green color to the DNA when UV light is shown on the agarose gel.
A. Reading DNA:
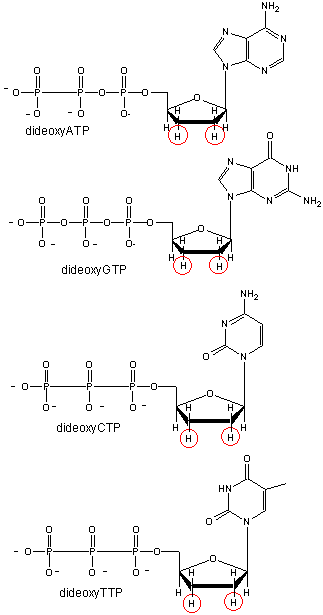
We will discuss one method of reading the sequence of DNA. This method, developed by Sanger won him a second Nobel prize. To sequence a single stranded piece of DNA, the complementary strand is synthesized. Four different reaction mixtures are set up. Each contain all 4 radioactive deoxynucleotides (dATP, dCTP, dGTP, dTTP) required for the reaction and DNA polymerase. In addition, dideoxyATP (ddATP) is added to one reaction tube The dATP and ddATP attach randomly to the growing 3' end of the complementary stranded. If ddATP is added no further nucleotides can be added after since its 3' end has an H and not a OH. That's why they call it dideoxy. The new chain is terminated.. If dATP is added, the chain will continue to grow until another A needs to be added. Hence a whole series of discreet fragments of DNA chains will be made, all terminated when ddATP was added. The same scenario occurs for the other 3 tubes, which contain dCTP and ddCTP, dTTP and ddTTP, and dGTP and ddGTP respectively. All the fragments made in each tube will be placed in separate lanes for electrophoresis, where the fragments will separate by size.
Didexoynucleotides
Figure: Didexoynucleotides
PROBLEM: You will pretend to sequence a
single stranded piece of DNA as shown below. The new nucleotides are added
by the enzyme DNA polymerase to the primer, GACT, in the 5' to 3' direction.
You will set up 4 reaction tubes, Each tube contains all the dXTP's. In
addition, add ddATP to tube 1, ddTTP to tube 2, ddCTP to tube 3, and ddGTP
to tube 4. For each separate reaction mixture, determine all the possible
sequences made by writing the possible sequences on one of the unfinished
complementary sequences below. Cut the completed sequences from the page,
determine the size of the polynucleotide sequences made, and place them as
they would migrate (based on size) in the appropriate lane of a imaginary
gel which you have drawn on a piece of paper. Lane 1 will contain the
nucleotides made in tube 1, etc. Then draw lines under the positions of the
cutout nucleotides to represent DNA bands in the gel. Read the sequence of
the complementary DNA synthesized. Then write the sequence of the ssDNA that
was to be sequenced.
5' T C A A C G A T C T G A 3' (STAND TO SEQUENCE)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
3' G A C T 5' (primer)
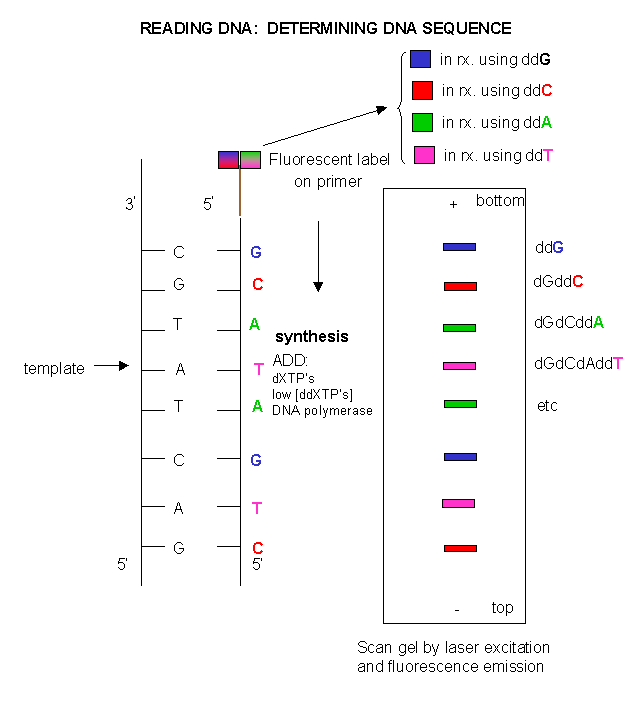
Since the DNA fragments have no detectable color, they can not be directly visualized in the gel. Alternative methods are used. In the one described above, radiolabeled ddXTP's where used. Once the sequencing gel is run, it can be dried and the bands visualized by radioautography (also called autoradiography). A place of x-ray film is placed over the dried gel in a dark environment. The radiolabeled bands will emit radiation which will expose the x-ray film directly over the bands. The film can be developed to detect the bands. In a newer technique, the primer can be labeled with a flourescent dye. If a different dye is used for each reaction mixture, all the reaction mixtures can be run in one lane of a gel. (Actually only one reaction mix containing all the ddXTP's together need be performed.) The gel can then be scanned by a laser, which detects fluorescence from the dyes, each at a different wavelength.
Figure: DNA sequencing using different fluorescent primers for each ddXTP reaction
One recent advance in sequencing allows for real-time determination of a sequence. The four deoxynucleotides are each labeled with a different fluorphore on the 5' phosphate (not the base as above). A tethered DNA polymerase elongates the DNA on a template, releasing the fluorophore into solution (i.e. the fluorophore is not incorporated into the DNA chain). The reaction takes place in a visualization chamber called a zero mode waveguide which is a cylindrical metallic chamber with a width of 70 nm and a volume of 20 zeptoliters (20 x 10-21 L). It sits on a glass support through which laser illumination of the sample is achieved. Given the small volume, non-incorporated fluorescently tagged deoxynucleotides diffuse in and out in the microsecond timescale. When a deoxynucleotide is incorporated into the DNA, its residence time is in the millisecond time scale. This allows for prolonged detection of fluorescence which give a high signal to noise ratio. Newer technology in which sequence is done by moving DNA through pores in membranes could bring sequencing down to $1000/genome or less.
![]() Animation
of Sanger Sequencing
Animation
of Sanger Sequencing
B. Writing DNA:
Oligonucleotide can be synthesized on a solid bead. By adding one nucleotide at a time, the sequence and length of the oligonucleotide can be controlled.
C. Copying DNA:
Several methods exists for copying a sequence of DNA millions of times. Most methods make use of plasmids (which are found in bacteria) and viruses (which can infect any cell). The DNA of the plasmid or virus is engineered to contain a copy of a specific DNA sequence of interest. The plasmid or virus is then reintroduced into the cell where amplification occurs.
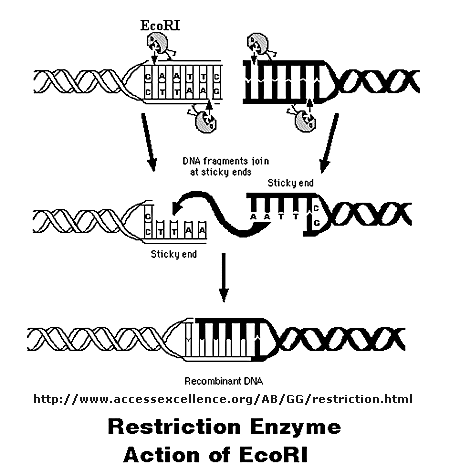
Initially, a DNA containing a gene or regulatory sequence of interest is cut at specific places with an enzyme called a restriction endonuclease, or restriction enzyme for short. The enzyme doesn't cleave DNA any old place, but rather at "restricted" places in the sequence, much as an endoprotease cleaves a protein after a given amino acid within a protein chain. Instead of cleaving one strand, as in proteins, the restriction endonuclease must cleave both strands of dsDNA. It can cut the strands cleanly to leave blunt ends, or in a staggered fashion, to leave small tails of ssDNA. Multiple such sites exist at random in the genome. The gene of interest must be flanked on either side by such a sequence. The same enzyme is used to cleave the plasmid or virus DNA.
Figure: Cleaving DNA with the Restriction Enzyme EcoR1
The foreign fragment of DNA can then be added to the plasmid or viral DNA as shown to make a recombinant DNA molecule. This technique of DNA cloning is the basis for the entire field of recombinant DNA technology.
Figure: Cloning a Restriction Fragment into a Plasmid
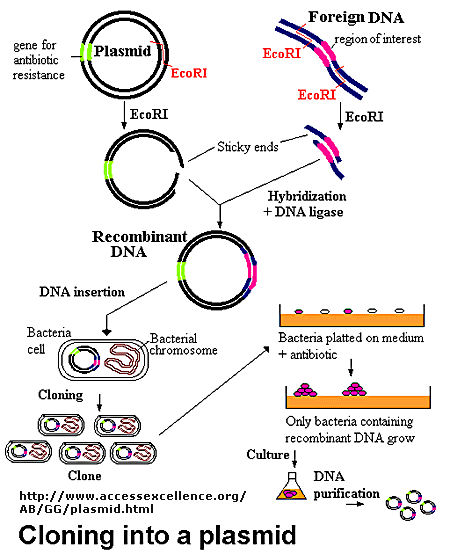
The plasmid can be added to bacteria, which take it up in a process called transformation. The plasmid can be replicated in the bacteria which will copy the DNA fragment of interest. Typically the plasmid carries a gene that can make the bacteria resistant to an antibiotic. Only bacteria that carry the plasmid (and presumably the insert) will grow. To isolate the desired fragment, the plasmids are isolated from bacteria, and cleaved with the same restriction enzyme to remove the desired fragment, after which it can be purified. In addition, the bacteria can be induced to express the protein from the foreign gene. In lab 4, we will transform bacteria with a plasmid containing the gene for human adipoctye acid phosphatase beta, HAAP-B, and induce expression of the gene.
A similar method can be used to copy DNA in which the foreign fragment is recombined with the DNA of bacteriophage , a virus which infects bacteria like E. Coli. The recombinant DNA can be packaged into actual viruses, as shown below. When the virus infects the bacteria, it instructs the cells to make millions of new viruses, hence copying the foreign fragment of interest.
Sometimes, "cloning" or copying a fragment of DNA is not what an investigator really wants. If the genomic DNA comes from a human cell, for instance, the gene will contain introns. If you put this DNA into a plasmid or bacteriophage, the introns go with it. Bacteria can replicate this DNA, but often one wants not to just copy (amplify) the DNA but also transcribe it into RNA and then translate it into protein. Bacteria, however, can not splice out the intron RNA, so mature mRNA can not be made. If one could clone into the bacteria DNA without the introns, this problem would not exist. One such possible method exists in which you start with the actual mRNA for a protein of interest. In this technique, a dsDNA copy is made from a ss-mRNA molecule. Such dsDNA is called cDNA, for complementary or copy DNA. This can then be cloned into a plasmid or bacteriophage vector and amplified as described above.
COPYING DNA FROM A M-RNA - CONSTRUCTION OF A C-DNA LIBRARY - insert
In the mid 80's a new method was developed to copy (amplify) DNA in a test tube. It doesn't require a plasmid or a virus. It just requires a DNA fragment, some primers (small polynucleotides complementary to sections of DNA on each strand and straddling the section of DNA to be amplified. Just add to this mixture dATP, dCTP, dGTP, dTTP, and a heat stable DNA polymerase from the organism Thermophilus aquaticus (which lives in hot springs), and off you go. The mixture is first heated to a temperature which will cause the DsDNA strands to separate. The temperature is cooled allowing a large stoichimetric excess of the primers to anneal to the ssDNA. The heat stable Taq polymerase (from Thermophilus aquaticus) polymerizes DNA from the primers. The temperature is raised again, allowing dsDNA strand separation. On cooling the primers anneal again to the original and newly synthesized DNA from the last cycle and synthesis of DNA occurs again. This cycle is repeated as shown in the diagram. This chain reaction is called the polymerase chain reaction (PCR). The target DNA synthesized is amplified a million times in 20 cycles, or a billion times in 30 cycles, which can be done in a few hours.
Figure: Copying DNA in the test tube - the polymerase chain reaction (PCR)
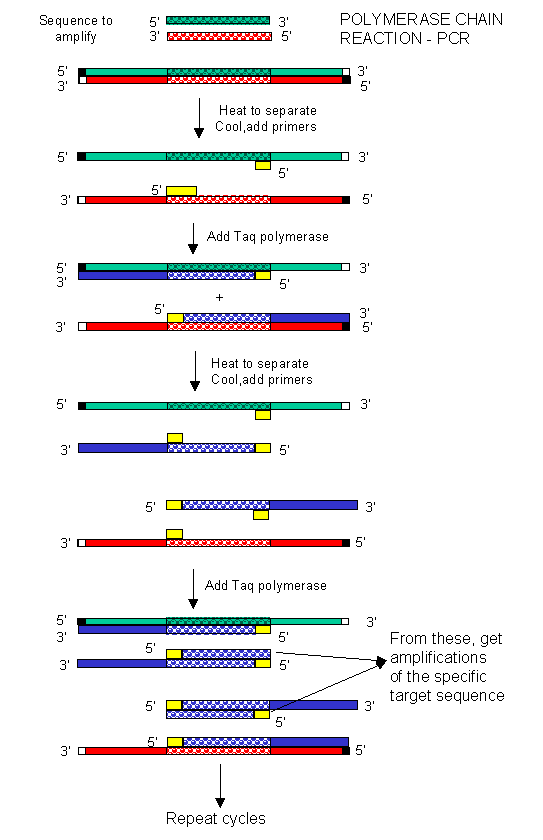
D. Editing DNA
During our studies of protein structure, we spent much time discussing how specific amino acids could be covalently modified to either identify the presence of the amino acid, or in an attempt to modify the activity of the protein. A newer and revolutionary technique has emerged in the last 15 years. Using recombinant DNA technology, the gene that encodes the protein can be altered at one or more nucleotide, in a way which would either change one or more amino acids, or add or delete one or more amino acids. This technique, called site-specific mutagenesis, is used extensively by protein chemist to determine the importance of a given amino acid in the folding, structure, and activity of a protein. The techniques is described in the diagram below;
Figure: Site Specific Mutagenesis
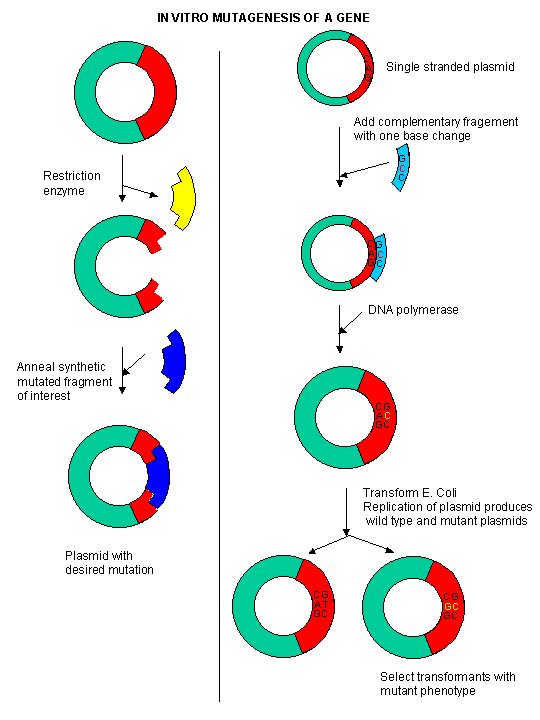
E. Searching DNA
Where on a chromosome is the gene that codes for a given protein? One way to find the gene is to synthesize a small oligonucleotide "probe" which is complementary to part of the actual DNA sequence of the gene (determined from previous experiments). Attach a fluorescent molecule to the DNA probe. Then take a cell preparation in which the chromosomes can be seen under the microscope. To the cell add base which unwinds the double stranded DNA helix, add the fluorescent probe to the cell, and allow double stranded DNA to reform. The fluorescent probe will bind to the chromosome at the site of the gene to which the DNA is complementary. Hybridization is the process whereby a single-stranded nucleotide sequence (the target) binds through H-bonds to another complementary nucleotide sequence (the probe).
What if you don't know the nucleotide sequence of the gene, but you know the amino acid sequence of the protein, as in the example shown below? From the genetic code table, you could predict the possible sequence of all possible RNA molecule which are complementary to the DNA in the gene. Since some of the amino acids have more than one codon, there are many possible sequences of DNA which could code for the protein fragment. The link below shows all possible corresponding mRNA sequences that could code for a short amino acid sequence. The 20 mer sequence of minimal degeneracy in the nucleotide sequence should be used as possible genomic probe .
F. COMPARING DNA
The DNA sequence of each individual must be different from every other individual in the world (with the exception of identical twins). The difference must be less than the difference between a human and a chimp, which are 98.5 % identical. Let us say that each of have DNA sequences that are 99.9 % identical as compared to some "normal human". Given that we have about 4 billion base pairs of DNA, that means we are all different in about 0.001 x 4,000,000,000 which is about 4 million base pairs different. This means that on the average we have one nucleotide difference for each 1000 base pairs of DNA. Some of these are in genes, but most are probably in between DNA, and many have been shown to be clustered in areas of highly repetitive DNA at the ends of chromosomes (called the telomeres) and in the middle (called the centromeres).
Now remember that their are restriction enzyme sites interspersed randomly along the DNA as well. If some of the differences in the DNA among individuals occurs within the sequences where the DNA is cleaved by restriction enzymes, then in some individuals a particular enzyme won't cleave at the usual site, but at a more distal site. Hence, the size of the restriction enzyme fragments should differ for each person. Each persons DNA, when cut by a battery of restriction enzymes, should give rise to a unique set of DNA fragments of sizes unique to that individual. Each persons DNA has a unique Restriction Fragment Length Polymorphism (RFLP). How could you detect such polymorphism?
You already know how to cut sample DNA with restriction enzymes, and then separate the fragments on an agarose gel. An additional step is required, however, since thousands of fragments could appear on the gel, which would be observed as one large continuous smear. If however, each fragment could be reacted with a set of small, radioactive DNA probes which are complementary to certain highly polymorphic sections of DNA (like teleomeric DNA) and then visualized, only a few sets of discrete bands would be observed in the agarose gel. These discrete bands would be different from the DNA bands seen in another individual's gene treated the same way. This technique is called Southern Blotting and works as shown below. DNA fragments are electrophoresed in an agarose gel. The ds DNA fragments are unwound by heating, and then a piece of nitrocellulose filter paper is placed on top of the gel. The DNA from the gel transfers to the filter paper. Then a small radioactive oligonucleotide probe, complementary to a polymorphic site on the DNA, is added to the paper. It binds only to the fragment containing DNA complementary to the probe. The filter paper is dried, and a piece of x-ray film is placed over the sheet. Also run on the gel, and transferred to the sheet, are a set of radioactive fragments (which are not complementary to the probe), which serve as a set of markers to ensure that the gel electrophoresis and transfer to the filter paper was correct. This technique is shown on the next page, along with a RFLP analysis from a particular family.
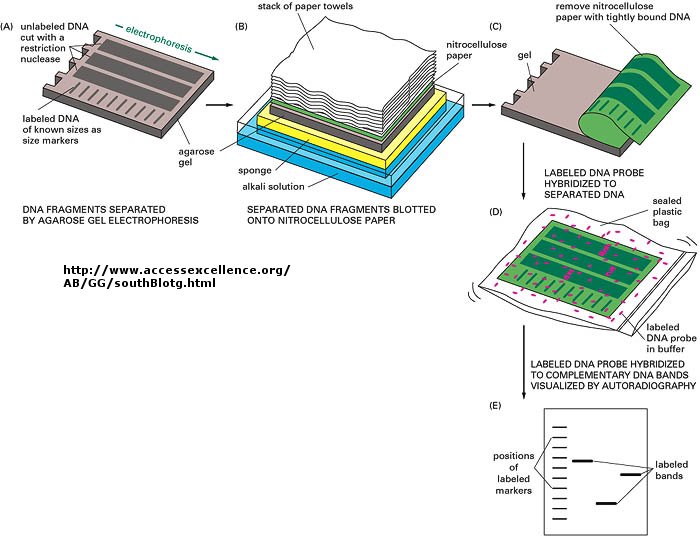{kind=link}
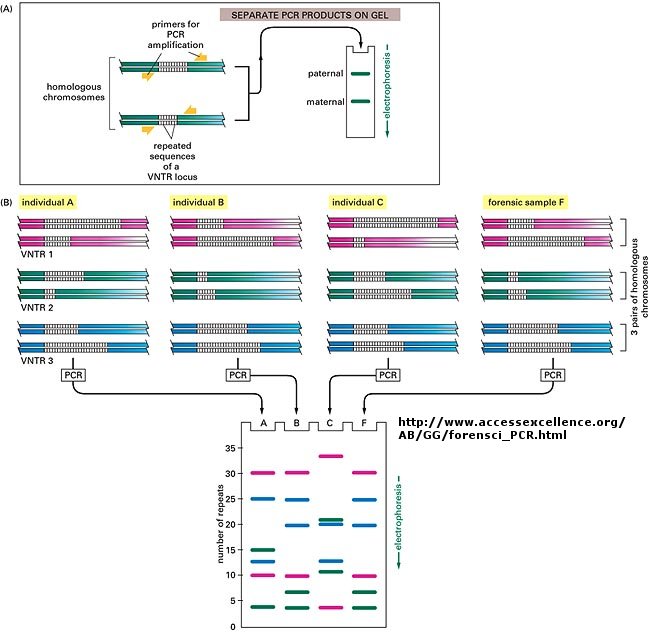
When this technique is used in forensic cases (such as the OJ Simpson trial) or in paternity cases, it is called DNA fingerprinting. With present techniques, investigators can state unequivocally that the odds of a particular pattern not belong to a suspect are in the range of one million to one. The x-ray film shown below is a copy of real forensic evidence obtained from a rape case. Shown are the Southern blot results from suspect 1, suspect 2, the victim, and the forensic evidence. Analyze the data.
Figure: SOUTHERN BLOT/PCR FORENSIC ANALYSIS - RESTRICTION FRAGMENT LENGTH POLYMORPHISM
Recent References
- Avise. Evolving Genomic Metaphors: A New Look at the Language of DNA. Science. 294, pg 86 (2001)
 Navigation
Navigation
Return to Biochemistry Online Table of Contents

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.