Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
D: PROTEIN FOLDING AND STABILITY
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/1/16
|
Learning Goals/Objectives for Chapter 2D: After class and this reading, students will be able to
|
D1. INTRODUCTION
We've seen many static images of lipids and their aggregates as well as proteins. To think about how proteins fold, we have to think dynamically. Luckily we have the tools of molecular dynamics (MD) at our fingertips which helps us imagine how these processes take place and concomitantly how to probe protein folding experimentally. View the following two MD simulations and compare the spontaenously formation of a micelle and the folding of a protein before we delve into the complex topic of protein folding and stability.
Given the number of possibly nonnative states, it is amazing that proteins fold to the native state at all, let alone in a reasonable time frame. Consider this greatly simplified view of protein folding for a protein containing 100 amino acids. If each amino acid can adopt only 3 possible conformations, the total number of conformations could be 3100 = 5 x 1047. Assuming that it would take 10-13s to change each conformation, the time required to "test" all conformations would be 5 x 1034s or 1027 years, longer than the age of the universe (14 x 109 yr). Yet the protein can fold within seconds. This paradox is called the Levinthal paradox, after Cyrus Levinthal.
Lubert Stryer (in his classic Biochemistry text), shows a way out of this dilemma by using an analogy of a monkey sitting at a typewriter, and typing this line out of Hamlet: "Me thinks it is like a weasel." Random typing would produce that line after 1040 keystrokes on average, but if the correct letters were maintained, the number of keystrokes would be in the realm of a few thousand. Proteins could fold more quickly if they retain native-like intermediates along the way. Also remember that much of conformation space is already restricted by allowed phi/psi angles. (Remember the blank areas in the Ramachandran plot?)
Before we study the classic experiment of protein folding conducted by Anfinsen, study the simpler analogy below:
Figure: Socks and protein folding
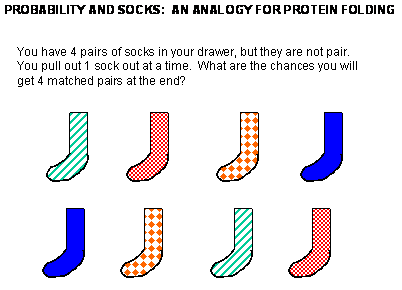
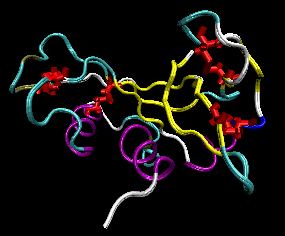
The classic experiment of Anfinsen has shown that, at least for some proteins, all the necessary and sufficient information required to direct the folding of a protein into the native state is present in the primary sequence of a protein. Anfinsen studied the in vitro (outside the cell, as opposed to in vivo, which is inside the cell, tissue, organ) folding of a single chain protein, RNase, which has 4 intrachain disulfide bonds.
Figure: RNase A with 4 Disulfide Bonds in red (image with VMD)
We have previously discussed how chemical agents (such as beta-mercaptoethanol, a disulfide reducing agent) can covalently interact with specific protein functional groups. Other substances can bind through complementary intermolecular forces to the active site or other cavities on the surface. Other reagents, like urea, acting through generalized solvent changes or nonspecific interactions with the protein, can alter protein folding. Anfinsen used two different reagents, 8 M urea and beta-mercaptoethanol, in combination to unfold, or denature, RNase to the nonnative or denatured state. He then removed the bME using dialysis, allowing the disulfides to reform. Next he removed the denaturing reagent, urea. To monitor if the protein was correctly refolded or renatured, he tested the activity of the protein compared to native protein. He found that the "refolded" protein retained only 1% of its initial activity. If, however, he added a catalytic amounts of bME, the protein soon retained 100% of its initial activity. For his work, he was awarded the Nobel Prize in Chemistry in 1972.
Figure: Anfinsen Experiment: Folding of RNase
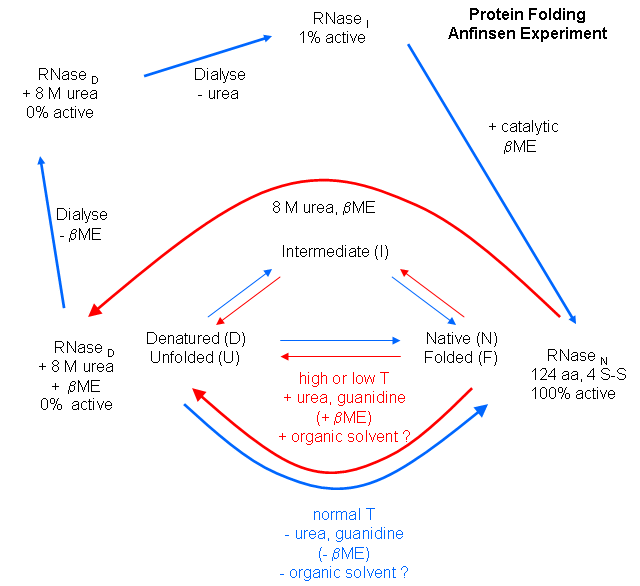
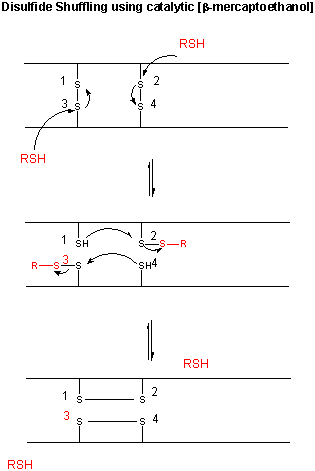
Figure: CATALYTIC SHUFFLING OF DISULFIDES WITH BETA-MERCAPTOETHANOL
![]() Jmol: Updated
RNase A
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
RNase A
Jmol14 (Java) |
JSMol (HTML5)
Scientists have investigated the folding of proteins both in
vitro and in vivo. In vitro experiments involve denaturing the protein with
urea, guanidine hydrochloride, or heat, then refolding the protein by
removing the perturbant (denaturing agent), using spectral techniques to
follow the process. In vivo experiments involve the study of intracellular
proteins that assist folding. The in vitro experiments involve unfolding the
native state and then refolding it, while the in vivo ones involve folding
of the newly synthesized protein. An understanding of protein folding
can not be separated from an understanding of protein stability, and an
understanding of the nature of the native and denatured state.
In studying protein folding and stability/structure of the native and denatured states, both equilibrium (thermodynamic) and timed (kinetic) measurements are made. Folding occurs in the ms to second range, which limits the ability to study the presence of intermediates in the process. Some clever methods have been developed to study intermediates in protein folding by trapping specific intermediate structures, and investigating their structure and stability in a "leisurely" fashion. Alternatively, intermediates can be studied as they occur using stop flow kinetics. In this technique, a protein under denaturing conditions is rapidly mixed with a solution containing no denaturant or protein by injecting both solutions into a mixer/cuvette using syringes. The denaturant in the protein solution is now diluted such that renaturation can occur. Spectral measurements can begin at once.
A diagram summarizing these methods is shown below. Study it in conjunction with the text which follows.
Figure: Kinetic and thermodynamic measurements of proteins stability and folding
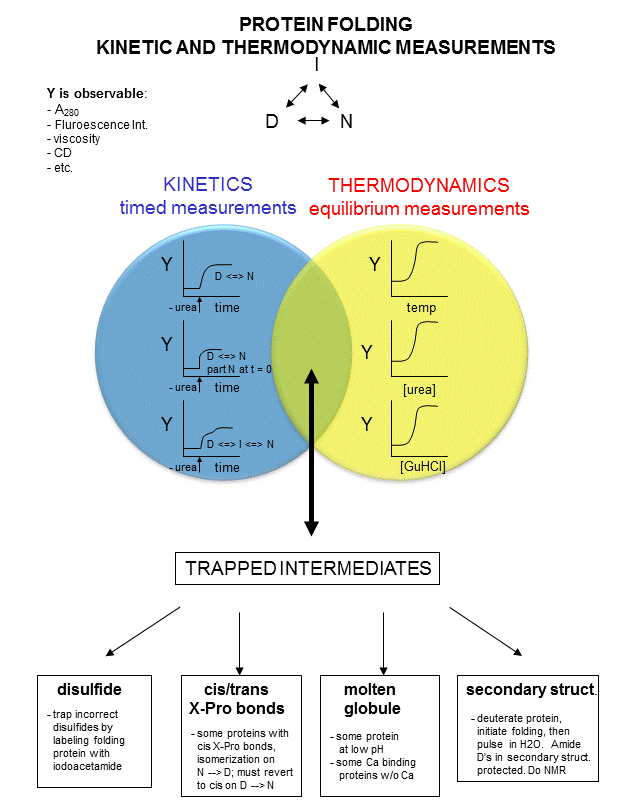
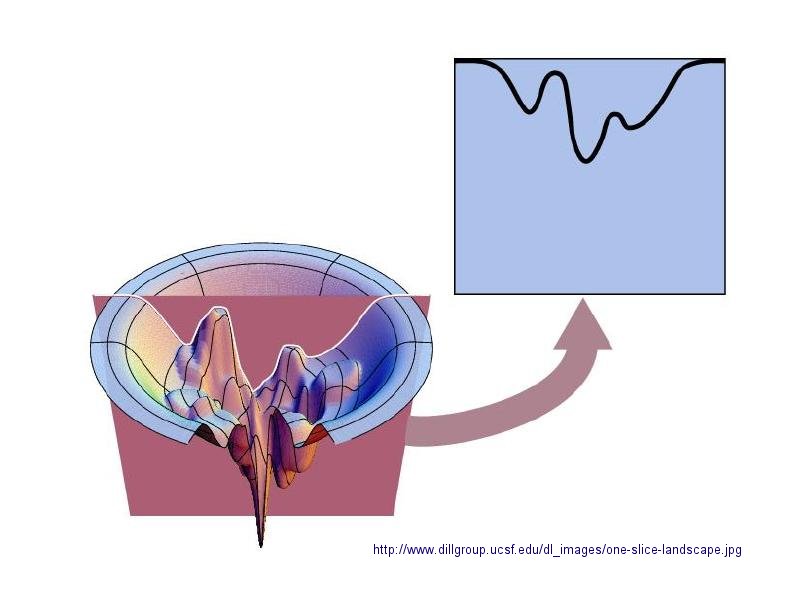
In considering the folding pathway, we will consider that the native protein represents the global energy minimum. All other states represent variations of the denatured state. Some, closer in energy to the native state, could be considered intermediates in the folding process. Instead of considering a folding "trajectory", consider protein folding occurring within a large folding landscape of free energy. Folding appears to proceed not by an obligatory pathway but a probabilistic or stochastic search of possible conformation. The free energy landscape must be shaped somewhat like a funnel such that a proteins could adopt a "reasonable" number of conformations which lead to the native state. Evolution has surely selected for sequences that can make it to that state. Localized secondary structure motifs (like a short alpha helix and beta turns) can form quickly (about 1 ms). Small proteins folding occurs, depending on the structure, over a wide time frame (ms to minutes). Mostly likely, a small number of amino acids coalesce into a core which nucleates folding into structures that are similar to the native state. Finally packing interactions collapse the structure into the native state.
In general, the more complex the fold of the backbone, the longer it takes the protein to fold. If complexity requires more interactions among distal regions of the polypeptide change, then the more complex the fold, the less probable that random interactions would lead to quick protein folding. The mechanisms of folding for larger proteins (greater than 100 amino acids) appear to proceed through intermediates, suggesting that different domains of the protein can fold independently.
Figure: Protein Folding Landscape: One View from Ken Dill
 Navigation
Navigation
Return to Chapter 2D: Protein Folding and Stability
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2D: Protein Folding and Stability

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.