Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
D: PROTEIN FOLDING AND STABILITY
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/1/16
|
Learning Goals/Objectives for Chapter 2D: After class and this reading, students will be able to
|
D5. Multiple Conformations from Same Sequence
1. Silent Single nucleotide polymorphisms (SNPs): For some amino acids, multiple triplet nucleotide sequences (codons) in the coding regions of a gene for a protein lead to the incorporation of the same amino acid in the protein sequence. Hence two proteins identical in amino acid sequence might have slightly different nucleotide sequences in the gene that encodes them. Such single nucleotide polymorphisms (SNPs) in coding regions were thought to have no effect on the tertiary structure and biological function of a protein if the single nucleotide variation did not lead to the insertion of a different amino acid into the growing peptide chain (i.e the codons were synonymous and the mutations presumably silent with no effect). Recently single nucleotide polymorphisms (SNPs) in the gene for the product of the MDR1 (multidrug resistance 1) gene, P-glycoprotein, was shown to result in a protein with different substrate specificity and inhibitor interactions, and hence a different 3D structure. One possible explanation for this observation is a difference in the rate of translation of the mRNA for this membrane protein. Different rates might lead to different intra- and intermolecular associations, which could lead to different final 3D structures as the protein cotranslationally folds and inserts into the membrane. This would especially be true if two possible structures where close enough in free energy but separated by a significant activation energy barrier, precluding simple conformational rearrangement of one conformation to another.
2. Metamorphic Proteins: In addition to prion proteins, it appears that many proteins can adopt more than one conformation under the same set of conditions. In contrast to prion proteins, however, in which the formation of the beta-structure variant is irreversible since the conformational change is associated with aggregation, many proteins can change conformations reversibly. Often, these changes do not appear to be associated only with binding interactions that trigger the change. Murzin has described proteins that change conformations on change of pH (viral glycoproteins), redox state (chloride channel), disulfide isomerization (lysozyme), and bound ligand (RNA polymerase as it initiates and then elongates the growing RNA polymer). He cites two proteins that appear to changes state without external signals. These include Mad2, in which the two conformers share extensive similarity, and Ltn10 (lymphotactin), in which they don't. One form of lymphotactin (Ltn 10) binds to similar lymphokine receptors, while the other (Ltn 40) binds to heparin. Folding kinetics may play a part in these examples as well, as proteins capable of folding to two conformers independently and quickly might prevent misfolding and aggregation that might occur if they had to completely unfold first before a conformational transition. Both Mad2 and Ltn10 alter conformation through transient formations of dimers, which facilitate conformational changes without widespread unfolding. Mutations in Ltn10 can cause the protein to adopt the Ltn40 conformation, Hence primordial "metamorphic" proteins could, by simple mutation, produce new protein functionalities.
3. Intrinsically Disordered Proteins (IDPs): Many examples of proteins that are partially or completely disordered but still retain biological function have been found. At first glance this might appear to be unexpected, since how could such a protein bind its natural ligand with specificity and selectivity to express its function? Of course one could postulate ligand binding would induce conformational changes necessary for function (such as catalysis) in an extreme example of an induced fit of a ligand compared to a "lock-and-key" fit. Decades ago, Linus Pauling predicted that antibodies, proteins that recognize foreign molecules (antigens), would bind loosely to the antigen, followed by a conformational change to form a more complementary and tighter fit. This was the easiest way to allow for a finite number of possible protein antibodies to bind a seemingly endless number of possible foreign molecules. This is indeed one method in which antibodies can recognize foreign antigens. Antibodies that bind to antigen with high affinity and hence high specificity more likely bind through a lock and key fit. (Pauling, however, didn't know that the genes that encode the proteins chains in antibodies are differentially spliced and subjected to enhanced mutational rates which allow the generation of incredible antibody diversity from a limited set of genes.)
It's been estimated that over half of all native proteins have regions (greater than 30 amino acids) that are disordered, and upwards of 20% of proteins are completely disordered. Regions of disorder are enriched in polar and charged side chains which follows since these might expected to assume many available conformations in aqueous solutions compared to sequences enriched in hydrophobic side chains, which would probably collapse into a compact core stabilized by the hydrophobic effect. Mutations in the disordered regions tend to preserve the disordered region, suggesting that the disordered region is advantageous for "future" function. In addition, mutations that cause a noncoding sequence to produce a coding one invariably produce disordered protein sequences. Disordered proteins tend to have regulatory properties and bind multiple ligands, in comparison to ordered one, which are involved in highly specific ligand binding necessary for catalysis and transport. The intracellular concentration of disordered proteins has also been shown to be lower than ordered proteins, possibly to prevent occurrences of inappropriate binding interactions mediated through hydrophobic interactions, for example. Processes to accomplish this include more rapid mRNA and protein degradation and slower translation of mRNA for disordered proteins. For a similar reason, misfolded proteins are targeted for degradation as well. Figure A below shows the mean net charge vs the mean hydrophobicity for 275 folded and 91 natively unfolded proteins. Figure B shows the relative amino acid composition of globular (ordered) proteins compared to regions of disorder greater than 10 amino acids in disordered proteins. The two different grey bars were obtained with two different versions of the software used to analyze the proteins. Again the graph shows an enrichment of hydrophilic amino acids in disordered proteins.
Figure: Characteristics of Intrinsically Disordered Proteins
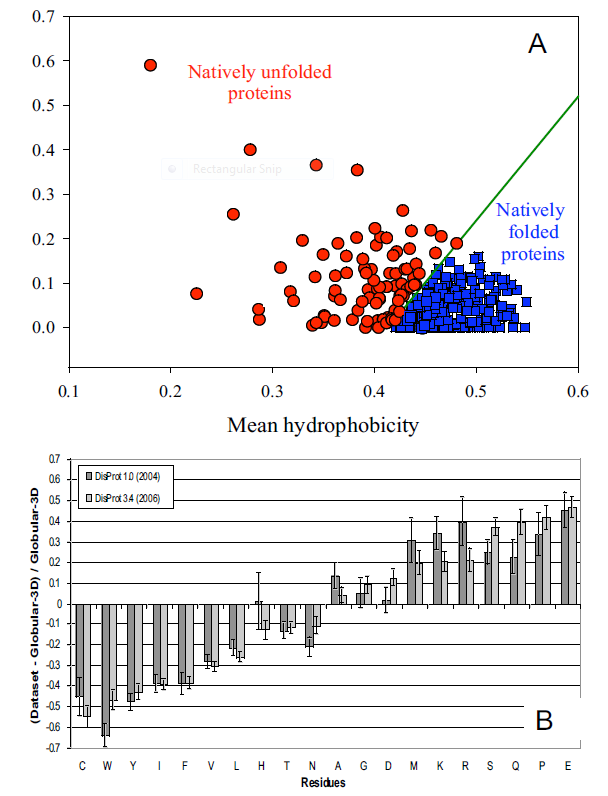
from open access journal: Dunker, A. et al. BMC Genomics 2008, 9(Suppl 2):S1 doi:10.1186/1471-2164-9-S2-S1
Many experimental methods can be used to detect disordered regions in proteins. Such regions are not resolved well in X-Ray crystal structures (have high B factors). NMR solution structures would show multiple, and differing conformations. CD spectroscopy likewise would show ill-defined secondary structure. In addition solution measurements of size (light scattering, centrifugation) would show larger size distributions for a given protein.
What types of proteins contain disorder? The above experimental and new computational methods have been developed to classify proteins as to their degree of disorder. There appears to be more IDPs in eukaryotes than in archea and prokaryotes. Many IDPS are involved in cell signaling processes (when external molecules signal cells to respond by proliferating, differentiating, dying, etc). Most appear to reside in the nucleus. The largest percentage of known IDPs bind to other proteins and also to DNA. These results suggest that IEPs are essential to protein function and probably confer significant advantages to eukaryotic cells as multiple functions can be elicited from the interaction of a single IEP (derived from a single gene) with different protein binding partners. This would greatly extend the effective genome size in humans, for examples, from around 25,000 with specified function, to many more. This doesn't even take into account the increase functionalities derived from post-translational chemical modifications.
We will discuss intrinsically disordered proteins further in Chapter 5. What is clear from recent finding is that protein structure is fluid and complex and our simple notions and words to denote proteins as either native or denatured are misguided and constrain our ideas about how protein structure elicits biological function. For example, what does the word "native" mean, if proteins exist in multiple states in vivo and in vitro simultaneously? Dunker et al (2001) have coined the concept "Protein Trinity" to move past the notion that a single protein folds to a single state which elicits a single function. Rather each of the states in the "trinity", the ordered, collapsed (or molten globule) and extended (random coil) coexist in the cell. Hence all can be considered "native" and all contribute to the function of the cell. A single IDP could bind to many different protein partners, each producing different final structures and functions. IDPs would also be more accessible and hence susceptible to proteolysis, which would lead to a simple mechanism to control their concentrations, an important way to regulate their biological activity. Their propensity to post-translational chemical modification would likewise lead to new types of biological regulation.
Figure: The Protein Trinity: Ordered, Collapsed and Extended States
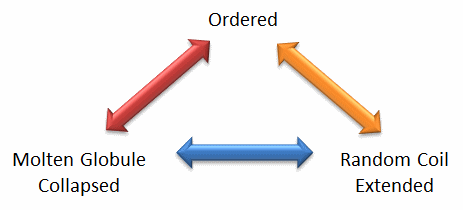
These ideas have profound ramifications for our understanding of the expression of cellular phenotype. In addition, a whole new world of drug target is available by finding drugs that modulate the transitions between ordered, collapsed and extended protein states. Likewise, side effects of drugs might be understood by investigating drug effects of these transitions in IDPs not initially targeted.
-
 PONDR
- Predictor of Naturally Occurring Disorder
PONDR
- Predictor of Naturally Occurring Disorder -
Database
of Protein Disorder
4. Catalysis by Molten Globule: A recent example (Bemporad) that a bacterial acylphosphatase has catalytic activity as a molten globule further questions our notions of structure and enzyme activity. In this example, substrate interaction did not induce global conformational changes in the protein. Molecular dynamics simulations showed that many partially disordered conformations of the protein are present, and the disorder involved the active site. However, parts of the protein are more ordered and form a "scaffold" which keeps the catalytic and substrate binding amino acids near enough that binding could engender conformational rearrangements at the active side and subsequent catalytic activity.
 Navigation
Navigation
Return to Chapter 2D: Protein Folding and Stability
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2D: Protein Folding and Stability

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.