Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
F: THERMODYNAMICS AND IMFs IN PROTEIN STABILITY
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/2/16
|
Learning Goals/Objectives for Chapter 2F: After class and this reading, students will be able to ...
|
F5. Additives and Their Interactions with Protein Surfaces
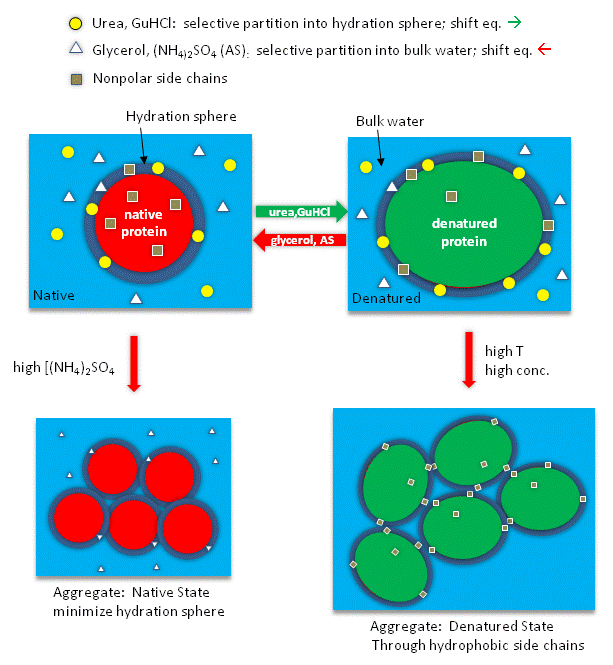
Additives to proteins that increase the stability of the folded state of the protein also tend to decrease their solubilities. These additives are excluded from the preferential water hydration sphere around the protein (negative binding of these agents). Denaturants in contrast tend to increase protein solubility and interact preferentially with the protein surface. In their presence, proteins respond by increasing their surface area by denaturation. For stabilizers, proteins try to minimize their surface area by staying "native" and aggregating to form a precipitate, both of which minimizes the surface area from which the stabilizing agent is excluded.
The main effect of dissolved ions on water structure has been thought to involve changes in H bonds (either enhancers/structure maker or inhibitors/structure breakers) which correlate with salting-in or salting-out effects of various ions. Many techniques have been used to study these interactions:
- viscosity: inferential information on structure
- diffraction (x-rays/neutrons): gives information on coordination number of solvation shell (static information)
- NMR: information on average relaxation of bulk and hydration sphere water around ions (dynamic information)
- molecular dynamics simulations: which gives insight into short but not long range interactions between ions and water.
Recent studies have provided conflicting support for the notion of structure makers/breakers. Omta et al. 2003 have used femtosecond mid-infrared pump-probe spectroscopy to study actual H-bonds between water molecules in salt solutions (Mg(ClO4)2, NaClO4, and Na2SO4). In pump-probe spectroscopy, a sample is excited with a short pulse (pump) and after a short time lag, with another pulse (probe), which interacts with the excited state. The linear-polarized infrared pulses (pump) were used to excite OH groups in solution, followed by a probe pulse which was polarized 45 degrees compared to the pump pulse. Only those excited OH groups that had rotated in the time interval between the pump and probe would be excited by the probe. Using this technique, the time frame for reorientation of the OH groups, which is related to the "stiffness" of the H bonds, can be determined. The salts had no effect on the rotational motion of bulk water outside of the the first hydration shell, which suggests that salts have no effects on the H bond networks in bulk water. Mg2+ ions are considered structure making, as the ions greatly increases the viscosity of water, brought about supposedly by increased H bonds among water molecules. This study does not support this model. Increased viscosity of Mg solutions must be attributed to those ions directly interacting with water molecules. The solution can be modeled as bulk water with small rigid spheres of ion + first hydration sphere. Clearly, much more experimental and theoretical work must be performed to gain structure insight into the role of salts on water structure. Until then, we will continue to try to understand the effects of different salt on water structure in descriptive term and with use of thermodynamic quantities.
Studies have shown that urea binds preferentially to the protein surface, and hence tends to increase the protein's surface area and hydrophobic exposure, and denature proteins. However, note in the figure below, that glycerol, a bigger polar but uncharged molecule, stabilizes the native state. This pair of uncharged additives have correspondingly similar effects on protein stability as do the charged guanidine HCl/ammonium sulfate pair.
Figure: How reagents might interact with the surface of the protein.
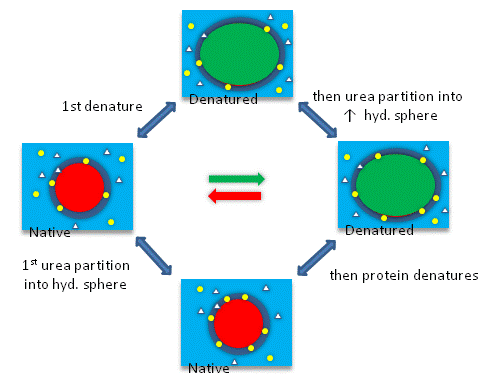
The next figures show such a thermodynamic cycle for urea denaturation of proteins
Figure: Thermodynamic cycle for urea denaturation of proteins
A graphical understanding of the type of sufrace changes that result from protein denaturation is given below.
Figure: Water Accessible Surface Area in Protein Unfolding
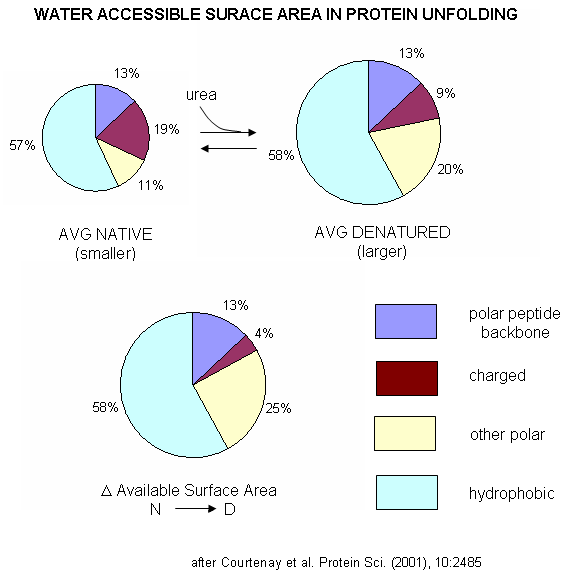
We will be discussing equilibria and how they may be shifted throughout this text.
A more detailed explanation of surface effects
and additives Courtenay, Capp, and Record (2001) used vapor phase osmometry to determine solution osmolality (Osm) and in turn preferential interaction coefficients (DGm3), from which KP values could be determined, using the formulas below, where a3 is the activity of component 3 - solute - in solution. (1 is solvent, 2 is protein, 3 is solute.) For example m1 = molality of bulk water = 55.5 mol/kg and b1 is the biopolymer hydration per square angstrom.. DGo obs = -RTlnKobs -(1/RT) DGo obs = lnKobs -(1/RT)( MDGo obs/Mlna3)T,P = MlnKobs/Mlna3) = DGm3 = (Mm3 /Mm2)T,P,m3 and DGm3 = (m3bulk(KP -1) b1 ASA)/m1 Using BSA as a protein solute, they
tested three destabilizers of native proteins: urea, guanidinium
HCl, and guanidinium thiocyanate. Graphs of osmolaity vs
destabilizer concentration (m3) showed almost linear increases in
both the presence and absence of BSA, but with lines that crossed
for the guanidium salts (lower osmolality in presence of BSA), which
indicated strong protein:solute interaction. Next, they determined, using vapor phase osmometry, the preferential interaction coefficients (from which KP values could be calculated) for BSA in the presence of increasing molal concentration (m3) of destabilizers. The values were always positive and increased with m3. KP values for the native protein were calculated to be 1.00 for KCl (control), 1.10 for urea, 1.30 for GuHCl, and 2.0 for GuSCN. For urea, previous calculations by the group showed the KP for urea and denatured proteins was the same as for native protein (1.10). Since it preferentially prefers to partition into the hydration sphere, and there is more hydration sphere in the larger denatured protein, urea drives protein unfolding. Using the guanidinium pair, they could resolve the KP values into that for the cation (GuH+) and the anion. For BSA, KP for GuH+ = 1.60 and 2.4 for thiocyanate. Similar calculations could be made of KP for the denatured state of BSA. The values for GuHCl and GuH+ were 1.16 (compared to 1.30 for the native protein) and 1.32 (compared to 1.60 for the native protein). Why does urea preferentially partition equally into the hydration spheres of native and denatured proteins, but GuHCl and GuH+ partitioned more into the native state? (Note however, that Kp for both N and D states were positive, leading to protein denaturation as with urea since the surface area and amount of hydration sphere greatly increases in the denatured state. ) Urea appears to partition selectively into the region near the peptide backbone and not charged or nonpolar surfaces. Theoretical and experimental work show that they are a constant percentage (13%) of the surface of a protein, whether in the native or denatured state, is composed of the peptide backbone. This make intuitive sense since the backbone extends over the entire length of the protein. However, the % of the surface with charged side chains decreases in the denatured state since then number of charged and polar side chains are a small fraction of the entire polypeptide backbone. The lower distribution of GuHCl and GuH+ into the hydration shell of denatured BSA can be accounted for by different distributions of surface polar and charged groups in the native and denatured state as shown in the figure below. For example, the percentage of charged groups on the surface of native vs denatured BSA is 29% vs 4%. It appears that GuHCl partitions into the hydration sphere near the peptide backbone and charged side chains. Urea appears to partition selectively into the region near the peptide backbone and not charged or nonpolar surfaces. This work gives a possible theoretical underpinning to the qualitative effects summarized in the Hofmeister series.
|
 Navigation
Navigation
Return to Chapter 2F: Thermodynamics and IMFs of Protein Stability
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2F: Thermodynamics and IMFs of Protein Stability

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.