Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
F: THERMODYNAMICS AND IMFs IN PROTEIN STABILITY
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/2/16
|
Learning Goals/Objectives for Chapter 2F: After class and this reading, students will be able to ...
|
F6. The Hydrophobic Effect and Change in Heat Capacity
Our understanding of hydrophobic interactions has changed dramatically in the last several years. This is not reflected in most textbooks. The hydrophobic effect mean different things to different people. Some refer to the transfer of nonpolar solutes/solvents to aqueous solution. Some refer to the same phenomena only if the effects have a unique temperature dependency. Other refer to the ordering of water around nonpolar residues. The most recent explanation centers around the unique temperature dependencies of the transfer reactions. Before we can understand it, here is an interesting bit of data. If you dissolve one mole of methane in hexane, the volume of 1 L of hexane changes 60 ml, but if done in water, the water volume changes 37 ml, indicating that water molecules pack more efficiently around nonpolar molecules then in its absence.
Let's now consider the thermodynamic aspects of the hydrophobic effect, as we did for micelle and bilayer formation. In a brief summary, we found the the free energy of transfer of an amphiphile from aqueous solution into a micelle, for example, was disfavored enthalpically (unexpectedly) but favored entropically (also unexpectedly until we included solvent in our model). These experiments were done at one temperature and gave us our first initial understanding of the hydrophobic effect. We will expand on this view by looking at the enthalpic and entropic contribution to the transfer of benzene into water as a function of temperature. This will lead us to a more modern view of the hydrophobic effect.
THERMODYNAMICS OF MIXING TWO SUBSTANCES
If you mix two substances A and B that aren't very soluble in each other, two opposing forces are relevant.
- The tendency to mix is driven by an increase in entropy.
- The mixing is usually opposed by enthalpy.
The later makes "intuitive" sense since you might expect that van der Waal forces between A-B might be less than those of A-A and B-B (i.e. the old adage "like dissolves like"). If AA and BB self interactions are stronger, A would not dissolve in B and vice/versa. You would also expect no significant changes in entropy and enthalpy as a function of temperature in this ideal mixing. See the graph below.
The most modern understanding of the hydrophobic interactions shows that we have mixing of A and B, but with a unique temperature dependency for the value of the change in entropy and enthalpies. At room temperature, if one corrects the entropy changes for effects due just to mixing, the "excess" entropy is what principally opposes taking a nonpolar molecule into water. Enthalpy changes are small. We have modeled this effecting using structured water around the nonpolar residues. Remember our discussions of micelle and liposome formation? We will further our understanding of the hydrophobic effect by studying benzene solubility in water.
TRANSFER OF BENZENE TO WATER AS A FUNCTION OF TEMPERATURE: A MODEL SYSTEM
Before we discus entropy and enthalpy changes accompanying protein folding/unfolding, let's try to learn about the thermodynamic aspects governing benzene solubility in water. What happens to benzene solubility in water and the corresponding thermodynamic parameters as you raise the temperature? The graph below shows the change in G, H and -TS for taking benzene from pure benzene to water. This is real data.
Figure: change in G, H and -TS for taking benzene from pure benzene to water.
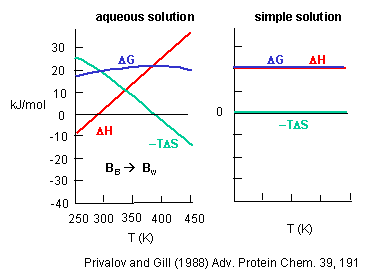
Notice the temperature dependency of these thermodynamic variables. As temperature increases, -TDS becomes more negative and hence drives benzene into water. (Remember D G = D H - TDS.) This seems to contradict our understanding of entropy effects in micelle and bilayer formation that we learned earlier when the DS > 0. Those studies on micelle and bilayer formation were done at one temperature. What about the structuring of water around the nonpolar group (decreasing the number of available microstates) hindering the process? Rationalize it this way. As the temperature is raised, available positional and thermal entropy of water increase significantly. It would seem logical that to then put a nonpolar residue into this system of water would become easier than putting it into more structure water (characterized by fewer microstates and lower positional and thermal entropy) at a lower temperature! (Remember from our review of thermodynamics that If the Tsurr is high, a given heat transfer to or from the surroundings will have a smaller effect on the ΔSsurr; conversely, if the Tsurr is low, the effect on ΔSsurr will be greater. Atkins, in his recent General Chemistry, uses the analogy of the effect of a sneeze in library compared to in a crowded street; The new American Chemistry General Chemistry text uses the analogy of giving $5 to a friend with $1000 compared to one who has just $10.)
But look at the other temperature anomaly. It becomes increasingly difficult from an enthalpic point of view to put benzene in water. We saw with micelle experiments that putting a hydrophobe into a micelle was disfavored enthalpically (and hence favored enthalpically to put it into water). At a high temperature, -TDS becomes zero, and their is no entropic barrier to putting benzene into water. The barrier is completely enthalpic. This is why a more nuanced definition of the hydrophobic effect has emerged.
If you sum DH and -TDS at each temperature, you get the curve shown for the total DG to take benzene from pure benzene to water. Notice that it is always positive, so it is always disfavored. The DG function is curved. It increases at low temperatures, and decreases at very high temperatures, implying that there will be a temperature at which there is a minimum solubility of benzene in water (a maximum in the positive DG). The minimum solubility of benzene (the max. positive DG) occurs when dG/dT = 0. Now remember from physical chemistry that
dG = VdP - SdT, so that dG/dT = V dP/dT - S = -S.
Therefore, dG/dT = 0 occurs when S = 0, and the maximum aversion is driven by enthalpy. This is at variance with the view that water ordering is the principal feature of the aversion of nonpolar residues for water.
Derivation for dG=VdP-SdT (for just PV work)

Let's review some more thermodynamics that the physical chemists in the crowd should remember. Even General Chemistry students should to a degree (a pun). (Molar) heat capacity, Cp, is defined as the heat required to raise the temperature of a mole of a substance 1oC. It has units of kJ (or kcal)/o mol. Look at the slope of the enthalpy curve as a function of temperature. It has units of kJ/o mol or of heat capacity and is given by:
Cp = dH/dT.
(Earlier, we studied Cp vs T curves for the phases transitions of water and for the gel/liquid crystalline phase transitions of phospholipid vesicles
Figure: Phase transitions of water
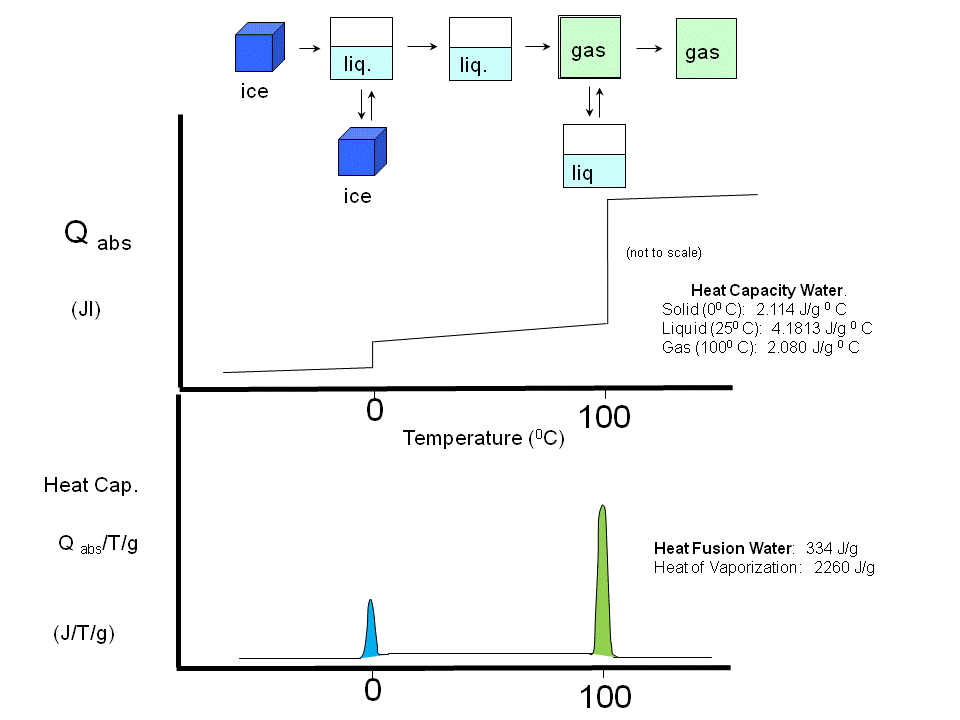
Figure: gel/liquid crystalline phase transitions of phospholipid vesicles
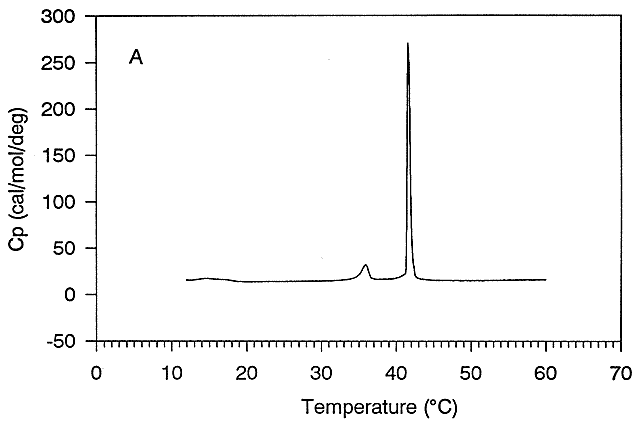
Figure: Thermal Denaturation of a protein using differential scanning calorimetry
Here is yet another equation for heat capacity that you derived in physical chemistry:
Cp = dH/dT= TdS/dT.
The last equality stems from Maxwell's relationships which Physical Chemistry students should remember. By analogy,
DCp = d(DH)/dT = Td(DS)/dT
which is POSITIVE in the benzene graphs above. THIS POSITIVE DCp OBSERVED WHEN A HYDROPHOBIC GROUP IS TRANSFERRED TO WATER IS THE SIGNATURE FOR OUR NEW DEFINITION OF THE HYDROPHOBIC EFFECT!
A positive DCp occurs when DH and DS are dependent on temperature, which is observed when a hydrophobe is transfer from a more nonpolar environment to water. Likewise, a negative DCp is observed when hydrophobes in water are transferred to a more nonpolar environment. Look at the graph showing the heat capacity of proteins vs temperature obtained using differential scanning calorimetry, as we previously studied with lipids.
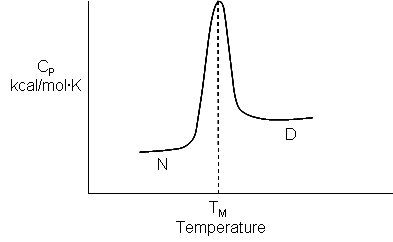
Figure: heat capacity of proteins vs temperature
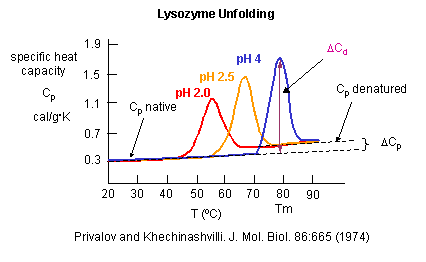
As the protein is heated, it reaches a temperature at which a large amount of heat is suddenly absorbed, as the protein unfolds. The area under the curve represents the heat absorbed on denaturation. The temperature at the midpoint is the Tm of the protein. (Why would the Tm be dependent on the pH of the solution?) Notice that there are two DCp's evident. One I will call DCd is associated with the actual denaturation process and is analogous to the change in heat capacity observed in phase changes, such as solid to liquid water. The other is D Cp which is the difference in heat capacity between the denaturated and native state. As was the case for the transfer of benzene to water, the DCp for protein denaturation is also positive, suggesting that in protein denaturation, hydrophobes are transferred from the interior of the protein to water.
Figure: Determination of DH for Protein Denaturation by Differential Scanning Calorimetry
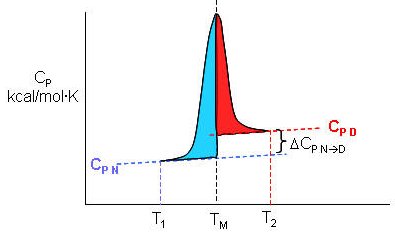
The following equations can be derived to help calculate the calorimetric enthalpy changes (DHcal = DHd) of denaturation from differential scanning calorimetry.
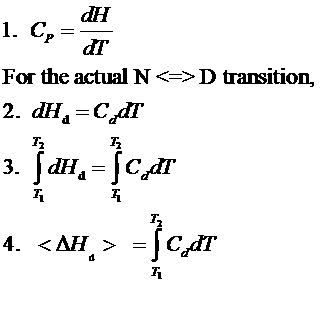
where <DHd> is the average enthalpy change that occurs on denaturation which is represented by the blue and red shaded areas in the curve above (between T1 and T2). Compare this to the van't Hoff enthalpy discussed in Laboratory Determination of DGo for Protein Folding/Unfolding.
For the calorimetric determination of DCd and DHd, the values are temperature dependent, and no assumption about the temperature independence need be made, as in the case of the DHvhoft .
What is the molecular basis for this large heat capacity change of transfer for benzene. One can show that the Cp is also proportional to the surface area of the nonpolar solute. At room temperature, water molecules surrounding the nonpolar residue are low in energy (lots of H bonds) and low in entropy (thermal and positional , fewer available microstates). As the temperature is raised, water populates higher energy states (fewer H bonds) and higher entropy (thermal and positional , more available microstates. The increase in temperature causes "melting" of surrounding water structure in so far as energy and entropy are concerned. The two different energetic states of water provide an energy storage mechanism.
Consider a slightly different explanation. Water molecules form an "iceberg"-like cage of water around nonpolar molecules, which is often called a clathrate. The water is fully H-bonded (to itself, not to the nonpolar molecule) in a fashion analogous to ice but the geometry of the H bonds is nonideal. This structuring of water decreases its entropy. With increasing temperature, the structured water "melts" which produces the large heat capacity of a solution of a nonpolar molecule in water, just as the actual melting of ice showed a large heat capacity. This large heat capacity is the signature thermodynamic features of the solution of a nonpolar molecule in water.
Heat capacity also had special significance in that
Cp = dH/dT = TdS/dT = -t2d2G/dT2
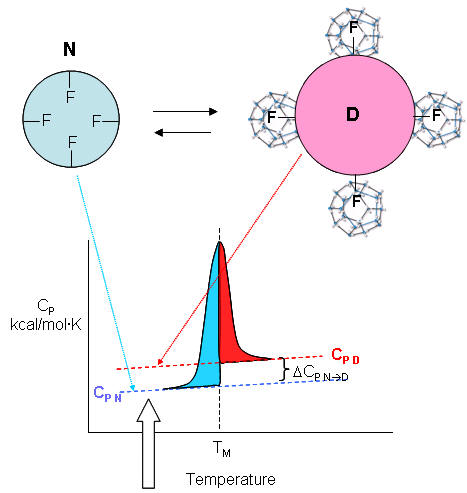
| A molecular interpretation of the "hydrophobic effect" It's still hard to get a good molecular interpretation of the meaning of DCp. The diagram below helps to directly relate the two. The smaller, more compact native state, with buried Phe (F) side chains denatures to the more open D state with exposed F side chains. Since these are nonpolar, we can envision a "clathrate" or cage of ordered water around them. I've extrapolated the heat capacity curves for both the native and denatured state into region where T < Tm (even though there is very little denatured state in that region). Now the "caged" water around the exposed F in the D state is low energy due to the "ice-like" H bond network. More heat would be absorbed (as the temperature is increased) to break up that cage compared to the same amount of heat applied to the N state. Hence, Cp D > Cp N. Is there a more quantitative description of the ordered water than a cage? Sharp et al investigated the "structure" of the caged water around nonpolar and polar molecules in a theoretical analysis supported by molecular dynamics (Monte Carlo) simulations. The average bond angles and lengths of water-water H bonds in the first hydration sphere around a nonpolar molecule like benzene decreased , but increased for polar ones such as potassium ions. The average changes noted arose from two types of H bonds compared to bulk water, those that were shorter and more linear, and those that were longer and more bent. They stated that "nonpolar groups do not so much increase the ordering of water as decrease the disordering".
A recent review by Silverstein suggests that an immobile clathrate cage is not a good representation for water surrounding a hydrophobe. Although we like to envision molecular models that allows us to "explain" experimental thermodynamic findings, such models themselves should be subjected to rigorous experimentation. An alternative explanation hinges on water's small size (compared to other solvents), its tight packing and high density. Consider the density of water compared to more nonpolar liquid solvents as seen the table below. Table: Density of common solvents
Let's consider the density of water surrounding an exposed nonpolar. If we envision the surrounding water as a clathrate, we might assume it is "ice" like. So what are the physical properties of ice and water than might give us insight into the water surrounding a nonpolar molecule? Ice, of course, has a lower density than liquid water. This can't be simply explained by the number of H bonds since experimental evidence shows that ice has an average of 4 H bonds per water molecule compared with liquid water, with an average of 2.4. Experimental data also shows that to accommodate water molecules into a rigid network of interacting waters with tetrahedral symmetry, the H-O-H bond angle increases to 106 from 104.5. Liquid water molecules, with fewer packing constraints, can self organize to maximize packing and hence macroscopic density. Studies suggests that ten water molecules solvate a buried methyl group and infrared studies show that four of these have significant barriers to rotational diffusion, suggest they are effectively immobilized and hence "ice-like". Silverstein suggests then that the water surrounding a nonpolar group on solution should be consider in a dynamic sense with some immobilized (as in ice) and the remaining more fluid-like. |
 Navigation
Navigation
Return to Chapter 2F: Thermodynamics and IMFs of Protein Stability
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2F: Thermodynamics and IMFs of Protein Stability

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.