Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
G: PREDICTING PROTEIN PROPERTIES FROM SEQUENCES
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/9/16
|
Learning Goals/Objectives for Chapter 2G: After class and this reading, students will be able to:
|
G2. Prediction of Secondary Structure
As we have seen previously, amino acids vary in their propensity to be found in alpha helices, beta strands, or reverse turns (beta bends, beta turns). These difference can be rationalized from the structure of each amino acid, as described before.
Figure: Amino Acid Structure and propensity for secondary structure
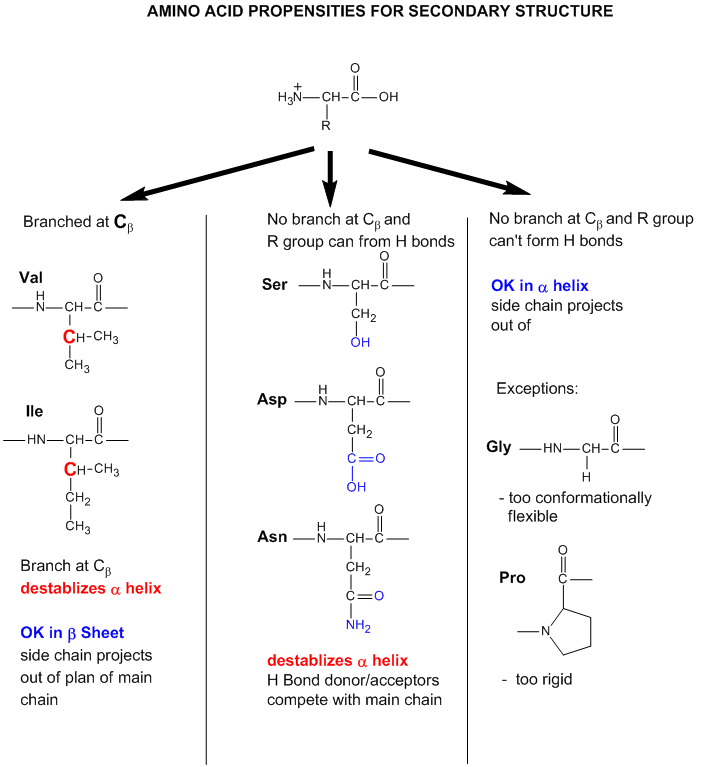
From the data bases, propensities can be calculated to determine the likelihood that a given amino acid will be in one of those structures. Glycine for example would have a high propensity to be in reverse turns, while Pro, a helix breaker, would have a low propensity to be in an alpha helix. A number is assigned to each amino acid for each category of secondary structure. High numbers favor the likelihood that that amino acid would be in that structure. One of the earliest propensity scales was from Chou-Fasman, where H indicates high propensity for secondary structure, h intermediate propensity, i is inhibitory, b is a intermediate breaker, and B is a significant breaker of secondary structure.
Chou-Fasman Amino Acid Propensities
| A.A. | Helix | Sheet | ||
| Designation | P | Designation | P | |
| Ala | H | 1.42 | i | 0.83 |
| Cys | i | 0.70 | h | 1.19 |
| Asp | I | 1.01 | B | 0.54 |
| Glu | H | 1.51 | B | 0.37 |
| Phe | h | 1.13 | h | 1.38 |
| Gly | B | 0.57 | b | 0.75 |
| His | I | 1.00 | h | 0.87 |
| Ile | h | 1.08 | H | 1.60 |
| Lys | h | 1.16 | b | 0.74 |
| Leu | H | 1.21 | h | 1.30 |
| Met | H | 1.45 | h | 1.05 |
| Asn | b | 0.67 | b | 0.89 |
| Pro | B | 0.57 | B | 0.55 |
| Gln | h | 1.11 | h | 1.10 |
| Arg | i | 0.98 | i | 0.93 |
| Ser | i | 0.77 | b | 0.75 |
| Thr | i | 0.83 | h | 1.19 |
| Val | h | 1.06 | H | 1.70 |
| Trp | h | 1.08 | h | 1.37 |
| Tyr | b | 0.69 | H | 1.47 |
Next a stretch or "window" of amino acids about 7 amino acids is taken, starting from the N-terminal of the protein. First the average alpha helical propensities for amino acids 1-7 are determined and assigned, let's say, to the middle (4th) amino acid in that sequence. Then alpha helical propensities for amino acids 2-8 (the next window) are averaged and assigned to the middle (5) amino acid in that range. The window slide down the protein sequence until all but the first and last few amino acids have an average value assigned to them. If a contiguous stretch of amino acids has high average propensity, they are probably in an alpha helix in the native protein. This process is repeated using beta strand and reverse turn propensities. The final assignments of most probably secondary structure are made. Of course this system was tested against proteins whose tertiary structure was known. See the results for secondary structure prediction for one protein. In this example, the average propensity for four contiguous amino acids is calculated (starting with amino acids 1-4, then amino acids 5-8, etc, and continuing to the end of the polypeptide). Next this process is repeated for contiguous stretches 2-5, 6-9, etc, and continuing to the end. The original Chou Fasman propensities have been updated using known protein structure to give better predictions.
- Chou Fasman Online Secondary Structure Predictor
Additional information about putative helices can be obtained by determining if they are amphiphilic (one side of the helix containing mostly hydrophobic side chains, with the opposite side containing polar or charged side chains. A helical wheel projection can be made. In this a circle is draw representing a downward cross-sectional view of the helix axis.
Figure: Helical wheel projection
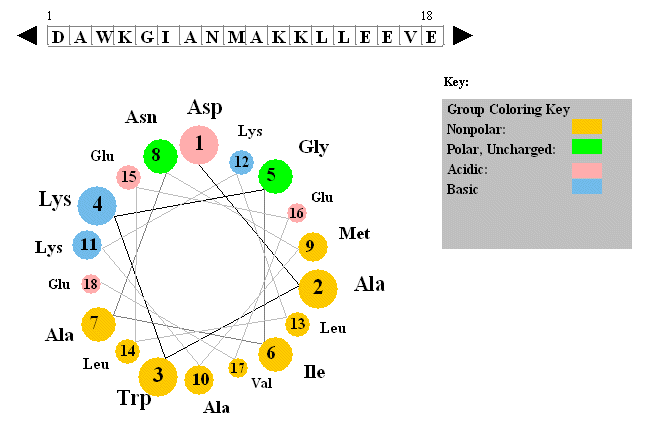
The side chains are placed on the outside of the circle, staggered in a fashion determined by the fact that there are 3.6 amino acids per turn of the helix. If one side of the wheel contains predominantly nonpolar side chains while the other side has polar side chains, the helix is amphiphilic. Imagine how such helices might be packed in a protein.
- Helical wheel predictor | Another Helical wheel predictor | Another
- Programs for Secondary Structure Prediction
 Navigation
Navigation
Return to Chapter 2G: Predicting Protein Properties from Sequences
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2G: Predicting Protein Property from Sequences

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.