Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
G: PREDICTING PROTEIN PROPERTIES FROM SEQUENCES
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/9/16
|
Learning Goals/Objectives for Chapter 2G: After class and this reading, students will be able to:
|
G6. Proteomics Problem Set 1
You will study a signal transduction protein and their interaction domains using a variety of web-based proteomics programs. For most of these programs you will need to input the amino acid sequence in FASTA format. Select a PDB code for a protein from the table at the end of this section. You could also use these programs to study any protein in the PDB.
Getting the FASTA sequence
1. First go to the PDB. Input the name
of your protein (which has an interaction domain) in the search box. Limit
the search to homo sapiens. Pick from the list of protein structure files
the most appropriate one. The example below is for the 2YYN pdb code.
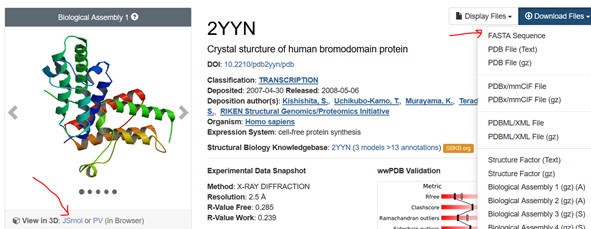
2. Select the Download Files dropdown and save the FASTA sequence to
your home directory. Download the file as a Wordpad. You might have to
remove recurring sections that don’t correspond to the single letter amino
acid sequence or identical sequences if the structure consists of identical
subunits To see if that might be the case, select JSmol (see figure above),
rotate the structure with your mouse to see if there are multiple chains,
and hover the mouse over the chains to see how the amino acids in that chain
are labeled. You might see [TRP]33A: for example, where A indicates a
separate A chain. Move to other chains. Then go to the Wordpad version of
the FASTA sequences. You can examine the chains to see if the chains are
identical. If so delete all but the first. See the above FASTA link for
help.
I. Prediction of Protein Properties from Sequence Data
Use
the following programs to gain information about your protein. Snip (with
snipping tool for example) and paste a bit of relevant info from each
program (using Snipping Tool) into this DOCX file and save it into the
folder and upload it into Sharepoint. Name the file
Lastname_LastName_FirstInitial_WebInteraction. If you have any problem with
any of the programs (lots of error messages), skip that particular program.
Several of them do the same type of analyzes. Compare the result. Snip and
paste sufficient content to show that you complete the question. Write
answers when asked to interpret the output.
a.
Sequence
Manipulation Suite: Determine the molecular weight of the protein.
b. Eukaryotic Linear Motif: Linear motifs are short, evolutionarily intrinsically
disordered section of regulatory proteins and provide low-affinity
interaction interfaces. These compact modules play central roles in
mediating every aspect of the regulatory functionality of the cell. They are
particularly prominent in mediating cell signaling, controlling protein
turnover and directing protein localization. The Eukaryotic Linear Motif
(ELM) provides the biological community with a comprehensive database of
known experimentally validated motifs, and an exploratory tool to discover
putative linear motifs in user-submitted protein sequences. Snip and paste
the top of the output that shows the IUPRED showing the disorder/order
graph.
c. TargetP
1.1 : predicts the subcellular location of eukaryotic
protein. Snip and paste the results. Interpret them based
on this link.
Where is your protein likely found?
d. NET-NES 1.1
Server:: predicts
leucine-rich nuclear export signals (NES) in eukaryotic protein This link
will help you
explain
the output. Does yours?
e. NLSdb --
Database of nuclear localization signals: Search for information on nuclear localization
signals (NLSs) and nuclear proteins. Select Query. Input the PDB code and
select NL. Does yours?
f. NetPhos
2.0 server: produces neural network
predictions for serine, threonine and tyrosine phosphorylation sites in
eukaryotic proteins. (other cool prediction programs from this site)
g.
TMPRED: The TMpred program makes a prediction of membrane-spanning regions
and their orientation. The algorithm is based on the statistical analysis of
TMbase, a database of naturally occurring transmembrane proteins. The
prediction is made using a combination of several weight-matrices for
scoring. Paste in your FASTA sequence but remove the header before running.
Does it have a transmembrane helix?
h.
TopPred 1.1 – Topology predictor
for membrane proteins at the Pasteur Institute. You will have to input your
email address. Paste in the entire FASTA file. Does it have transmembrane
helices? (Part of
Mobyle)
Save the first graph (PNG graphic file) of the
output, open it with Adobe Photoshop, and paste the image into your report.
Does the graph show alternating hydrophobic (+ values)/hydrophilic (-
values) sections consistent with transmembrane helices (for example you
would expect to see 7 hydrophobic stretches for GPCR)?
i. PFAM –
multiple analyses of Protein FAMilies. This program looks at the domain
organization of a protein sequence. Input the pdb code. When finished,
select “sequences” in the list below.

Then select the human sequence. Snip the resulting diagram and legend
showing the domain structure of the protein. You can also click on each
domain in the diagram to get more info on the domain. Does the protein have
the domain suggested in the beginning table?
j. Prosite: Input your
FASTA sequence in the Quick Scan mode. Select Exclude motifs with a high
probability of occurrence from the scan. Snip and Paste the Hits by Proifle
domain structure. Sometimes you might need a different code number, the
UniProtKB: Accession number. Get this from the PDB web page as shown below:
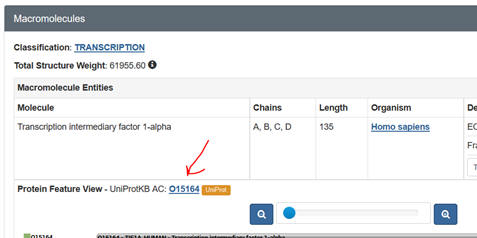
k.
eFindSite: is a ligand binding site prediction and virtual screening
algorithm that detects common ligand binding sites. Put in the PDB code and
then the pdb file you downloaded.
l.
eFindSitePPI: detects protein
binding sites and residues using meta-threading. It also predicts
interfacial geometry and specific interactions stabilizing protein-protein
complexes, such as hydrogen bonds, salt bridges, aromatic and hydrophobic
interactions
m.
NCBI Standard Protein BLAST: Input the FASTA file. The
output shows the domain and domain superfamily followed by other protein
sequences nearly identical to your protein. The results are graphical
followed by descriptive. Snip domain structure with the closest aligned
sequences. Then select under PDB structures the pdb code (example below
1xww).
You will see a window similar to below. Select under domain
1xwwwA00 (as an example). :
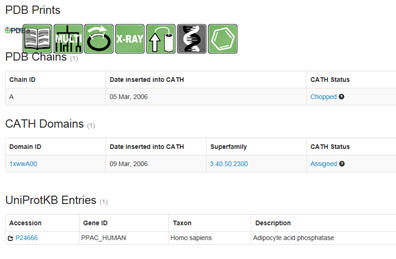
Then select the UniProtKB accession number. Confirm the many of the
predictions you made above.
n. Predict
Protein Open: Physiochemical properties of your protein.
You will have to provide your email address. When complete you can access
much of what you learned above by the links to the left under the Dashboard.
II. Visualizing Protein Interactions
It is important to be able to visualize the binding interactions between the targeted domain and the ligand (small molecule, PTM modified protein, protein or DNA). Here are some programs that allow that. Note: which programa you select will depend on if your protein is bound to a small ligand or to another protein or other macromolecule, in which case you need to explore protein interaction interfaces.
LIGAND:PROTEIN INTERACTIONS
Assignment: You will study the interaction of Protein Kinase C (PKC) with
the ligand phorbal ester, which is a mimic of the 2nd messenger
diacylglyceride with two programs: Ligand Explorer and Protein-Ligand
Interaction Profilers
a. Ligand Explorer is a Java-based program. It probably will NOT work
on a Mac running Safari. You will need the latest version of Java to run it.
Try the various computer labs around campus as well. Go to the PDB page for
your protein. After you input the pdb code, scroll down to Small Molecules
section in the middle of the displayed page for that complex. There are
links for both 2D and 3D visualization of the interactions. .
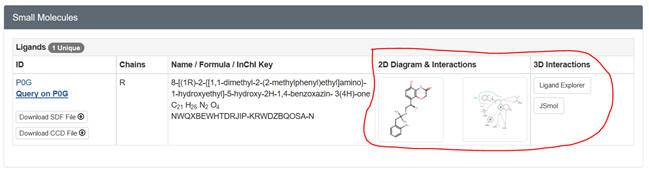
Select the 2D plot showing the interactions. The select Jsmol to see the
ligand with a binding surface) interacting with contact residues in the
protein. You can select a white background and toggle on and off H bonds.
SNIP and PASTE.
Now select Ligand Explorer for a more detailed view. Make
sure to select the correct ligand (see table below). You may be prompted to
allow pop-ups form the site. If so, allow it. You may have to reselect
Ligand Explorer again to start the program. Keep giving permissions and
following prompts until Ligand Explorer is open. Once launched, select open
this link in a new tab or window and instructions will open in a browser.
Use the mouse to help find the best view of the interactions.
Select in
turn hydrogen bond, hydrophobic, bridged H bond (mediated by a water
molecule) and metal interaction (shown on the left hand side. Select Label
Interactions by Distance. Take a cropped screenshot of each interaction (see
instructions below).
For the final rendering, move the toggle on the
Select Surfaces to opaque. Then change the distance in the best way to show
the cavity in which the ligand binds. Color by hydrophobicity which gives
two colors representing nonpolar and polar parts of cavity. Select solid
surfaces
b.
Protein-Ligand Interaction Profilers: Input the name of
your PDB file. After the run is complete, select SMALL MOLECULE and then the
appropriate ligand. You will get a 2D representation you can snip and paste.
Then select Pymol 3D view (first 5 computers in ASC 135). You will see an
interactive rendering of a small bound ligand and the protein residues it
contacts in the complex. You can get a
free student download of Pymol for
your own computer. Snip and Paste relevant info.
PROTEIN:MACROMOLECULE SURFACE INTERACTIONS
You will study
protein:protein interactions between a Src domain and small phospho-Tyr
peptide using InterProSurf and COCOMAPS.
a. InterProSurf: Reports numbers of surface and buried atoms for each chain, and areas for each residue deemed to be in the interface. Select PDB Complex in the top menu tabs and input your pdb file. This gives numerical data only. Snip and Paste relevant info.
b.
COCOMAPS: analyzes and visualizes interfaces in biological complexes
(such as protein-protein, protein-DNA and protein-RNA complexes). Input the
PDB file name and then the chains within the PDB file that you wish to see
the interaction surface. Put in the letter for one of the interacting chains
you selected into the first input box and the second letter into the second
box. Detailed results will appear in graphical and tabular form.

A great way to visualize the binding interface is to download the new .pdb and .pml files and open the pdb file in Pymol . Once the PDB file is opened in Pymol, select file -> run -> script_name.pml. Snip and Paste relevant info.
Table: Signaling Proteins for Analysis
|
Domains in Signaling Molecules |
||||
|
Domain |
Binding Target |
Cellular Process |
Example protein |
Pdb file |
|
Bromo |
Acetyl-Lys |
Chromatin reg. |
BRD4 |
2YYN |
|
C1 |
diacylglycerol |
Plasma memb recruitment |
Raf-1 |
3OMV |
|
C2 |
Phospholipid (Ca dependent) |
Membrane targeting, vesicle trafficking |
PRKCA |
3IW4 |
|
CARD |
Homotypic interactins |
apoptosis |
CRADD |
3CRD |
|
Chromo |
Methyl-Lys |
Chromo reg, gene txn |
CBX1 |
3F2U |
|
Death (DD) |
Homotypic inter. |
Apoptosis |
Fas |
3EZQ |
|
DED |
Homotypic inter. |
Apoptosis |
Caspase 8 |
1F9E |
|
DEP |
Memb, GPCRs |
Sig trans, prot trafficking |
Dsh human dishevelled 2 |
2REY |
|
GRIP |
Arf/Art G prot |
Golgi traffic |
Golgin-97 (Golga5) |
1R4A |
|
PDZ |
C-term peptide motifs |
Diverse, scaffolding |
PSD-95 Or discs large homolog 4 |
1L6O |
|
PH |
Phospholipids |
Membrane recuirt |
Akt |
1O6L 3CQW |
|
PTB |
Phosphor-Y |
Y kinase signaling |
Shc 1 SHC-transforming protein 1 |
1UEF 1irs europe |
|
RGS |
GTP binding pocket of Galpha |
Sig trans |
RGS4 |
1EZT |
|
SH2 |
phosphoY |
pY-signaling |
Src |
4U5W |
|
SH3 |
Pro-rich sequence |
Diverse, cytoskelet |
Src |
2PTK |
|
TIR |
Homo/Heterotypic |
Cytokine and immune |
TLR4 |
3VQ2 |
|
TRAF |
TNF signaling |
Cell survival |
TRAF-1 |
3ZJB |
|
VHL |
hydroxyPro |
ubiquitinylation |
VHL |
1VCB |
|
Protein Ligand and Protein Protein Interactions |
||||
|
Protein (PKC) :Ligand (phorbal ester mimic of 2nd
messenger diacylglyceride with Ligand Explorer and |
1PTR |
|||
|
Protein (Chain E-Src fragment) : Protein (Chain I –
phospho-peptide) with
COCOMAPS |
1QG1 |
|||
|
H-Ras-GppNHp bound to the Ras binding domain (RBD) of Raf
Kinase GppNHp binding with Ligand Explorer and
Protein-Ligand Interaction Profilers Protein (Ras, chain A):Protein (RBD-Raf, Chain B)
interactions with
COCOMAPS |
4G0N |
|||
 Navigation
Navigation
Return to Chapter 2G: Predicting Protein Properties from Sequences
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2G: Predicting Protein Property from Sequences

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.