PM9. Column Chromatography (Normal Phase)
Thin layer chromatography (TLC) can be used to separate many different mixtures. It is very flexible because several different compounds can be separated from each other in one experiment.
Practically speaking, TLC is often used only as an analytical tool rather than as a method of purification. It is used to quickly determine if a mixture is pure, how many compounds it may contain, and what combination of eluent and stationary phase can be used to separate the compounds. However, TLC often works best with a very small amount of material. Isolating useful amounts of compound sometimes requires other kinds of chromatography.
Column chromatography is another kind of liquid chromatography. It works just like TLC. The same stationary phase and the same mobile phase can be used.
Instead of spreading a thin layer of the stationary phase on a plate, the solid is packed into a long, glass column. Usually, the solid is slurried together with the solvent and poured into the column. Sometimes these columns are several inches wide and a few feet long. A large amount of material can be purified on a chromatography column.
Figure PM9.1. Introduction of a mixture to a chromatography column.
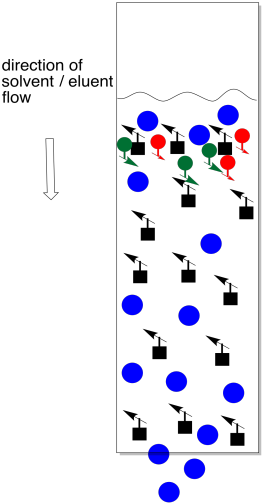
Instead of letting eluent wick up through the stationary phase, the solvent is poured into the top of the column and allowed to run through by gravity. The same factors of adhesion and solution in TLC apply here. If the same solid phase and liquid phase from TLC are used in a column, the compounds will elute through the column in the same order that they elute across a TLC plate.
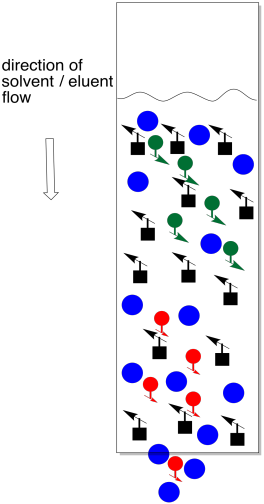
Figure PM9.2. Elution of a mixture on a chromatography column.
To collect the separated compounds, we just collect the solution as it drips out the end of the column. Normally we can't see the compounds, so we continuously collect fractions. That might mean collecting a mL of solution in one test tube, then a mL in the next test tube, and so on.
After collecting several fractions, we would have to check to see if they contained anything. Usually this is done by TLC. We carefully take a capillary tube and spot each fraction several times on a TLC plate. We take up some solution from the first fraction in a capillary tube, tap the capillary tube on the TLC plate where we want to spot the sample, and wait for the solvent to evaporate. Then we tap the capillary again, in the exact same spot. We will repeat this cycle a few times. What we are trying to do is build up enough compound on that spot so that we will be able to detect it (usually by shining a UV light on it). Of course, the fraction may be empty, so after the solvent evaporates there may nothing there anyway, but we don't know that until we check.
We then take the next fraction and do the same thing a little bit further along the TLC plate, and the same with the fraction after that. Eventually we have a row of spots, each corresponding to a different fraction. When we elute the TLC plate, we would ideally like to see fractions containing just one spot, at an Rf value corresponding to one of the components of the original mixture.
We collect any fractions that contain pure compound and evaporate them to obtain the isolated compound.

Figure PM9.3. Checking chromatography fractions via TLC.
That's the basic idea of a chromatography column, but let's look at a few practical details.
The first thing to think about is how we set up the column. Earlier we said that we load the stationary phase as a slurry, mixed together with the mobile phase already. Alternatively, sometimes the stationary phase is carefully poured into a column already full of the mobile phase. It would be a whole lot easier to just add the solid, stationary phase first, then pour in the mobile phase. That approach comes with its own problems.
The trouble is, stationary phases such as silica usually swell when they come in contact with the mobile phase. If you pour some silica into a column, then add solvent, the grains of silica puff up and get a whole lot more crowded. They get so crowded that there isn't much room for anything to move between them. If you have a couple of months to sit around and wait for your compound to elute through the column, this method may be right for you. Otherwise, don't try it.
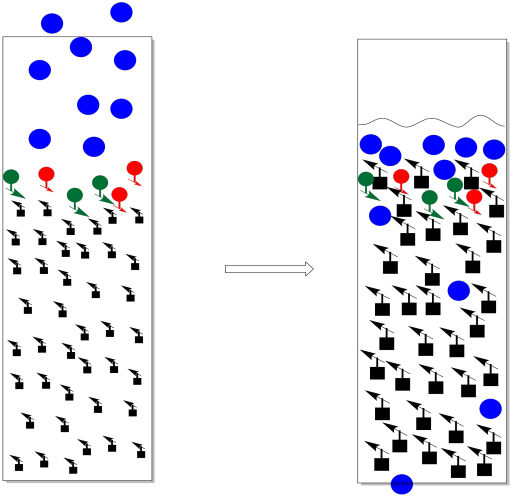
Figure PM9.4. Swelling of the stationary phase slows down a chromatography column.
As a general rule, we want the stationary phase to always be covered in solvent. If we let the stationary phase get exposed to air (if we let it run dry and don't replace the solvent), the opposite of swelling happens. The stationary phase shrinks. When it shrinks, it gets cracks in it. Maybe we notice the problem and add more solvent, but by then it may be too late. Instead of moving through the startionary phase like it should, everything just rushes through the cracks. There is no opportunity for any of the compounds in the mixture to separate from each other.
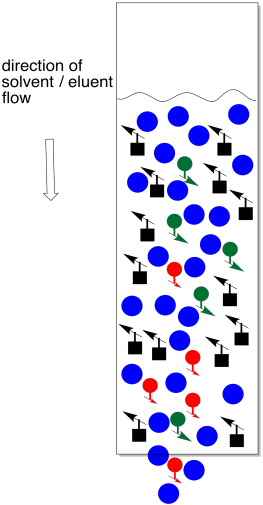
Figure PM9.5. A crack has developed in this chromatography column.
On the other hand, you can go oveboard with solvent. This is a particular problem at the beginning, when you are introducing the mixture to be separated. What you want to do at this point is let the eluent fall exactly to the same level as the top of the stationary phase, but no further. If it falls further, the column dries out. On the other hand, if you don't let it fall far enough, you have an equally big problem.
Usually the sample is introduced as a concentrated solution in a little bit of the eluent. Sometimes it is dried onto a little bit of stationary phase by making a mix of sample, stationary phase, and a solvent, then evaporating the solvent. Either way, the idea is to get the sample in a thin layer at the top of the column. We want a thin layer because chromatography is really a race and we want all of the molecules to be at the same starting line; we can't have anybody getting head start.
The trouble with adding too much solvent at this point is that some of the sample will dissolve and move backward into the eluent. We no longer have a thin layer of sample. Not everybody is at the same starting line. It now becomes almost impossible to separate the mixture.
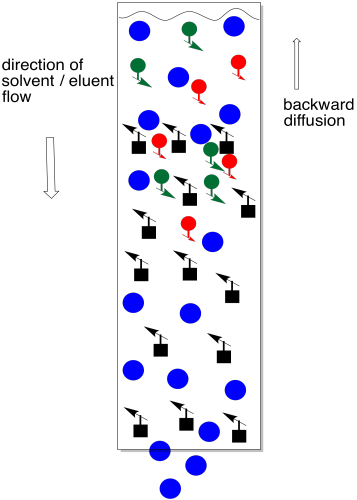
Figure PM9.6. Too much solvent at the beginning of a chromatography column.
Instead, we add a small amount of solvent and let it drop again to the top of the stationary phase. Repeat that a couple of times (three is a magic number in the lab). At this point, we have probably drawn the compounds down into the column far enough that there is none left on the top of the stationary phase. We can safely add more solvent without washing material backward.
Eventually, you will want to add as much eluent as you can, because the column operates under gravity. The more eluent you have above the column, the more weight pushes down to move things through the column. The column speeds up.
Sometimes, instead of letting the eluent run through the column via gravity, the eluent can be pushed through more quickly using an inert gas or an air pump. This method is called flash chromatography, and it requires some special equipment. There is sometimes a trade-off between quality of separation and the time it takes to run the column, though.
Problem PM9.1.
Why might there be less separation between two compounds if they both move through the column faster?
Problem PM9.2.
Suppose you are in the middle of separating a mixture on a chromatography column when you remember that you left the oven on in your apartment. Your apartment is a half hour from lab. You turn off the stopcock at the bottom of the column so that the eluent stops flowing. When you get back, you finish the column. You find that you didn't get very good separation between the compounds. What happened?
Problem PM9.3.
a) Suppose you are working with a chromatography column that can hold about 20 mL of solvent. You know that a sample is usually dissolved and then poured onto the top of the column before eluting with solvent. You dissolve your sample in 20 mL of solvent and proceed with the experiment. You get very poor results. What went wrong?
b) Meanwhile, the annoyingly perfect student in the next hood dissolves her sample in 1 mL of solvent and runs her column. She gets three pure compounds at the end and the instructor immediately gives her an A in the course. What did she do right?
Problem PM9.4.
Silica and alumina are not the only possible solid phases. For example, a C18 column contains beads that have 18-carbon chains attached to them. A C18 column is an example of a "reverse phase" column, which are often used with solvents such as water, methanol or acetonitrile. In a normal column, the stationary phase is more polar than the mobile phase; is that true in a reverse phase column?
This site is written and maintained by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (with contributions from other authors as noted). It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation:
Back to Structure & Reactivity