Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
H: PROTEIN AGGREGATES AND DISEASE
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 03/10/16
|
Learning Goals/Objectives for Chapter 2H: After class and this reading, students will be able to
|
H1. Protein Aggregation
We have seen that protein aggregates complicate the lives of people who study protein folding in vitro and who try to express human proteins in prokaryotes like E. Coli in vivo. Instead of viewing these aggregates as junk, some now study them avidly. It turns out that these aggregates are not as non-specific as earlier believed. In addition, an understanding of how and when they form will give us clues into the etiology and treatment of some of the most debilitating and feared diseases. Much of this review is based on the following reference: Taubes, G. Misfolding the Way to Disease, G. Science, 271, 1493-1495 (1996)
Clues Showing the Specificity of Aggregate Formation
- 1970's: It was shown that chymotrypsinogen could not be folded in vitro without aggregates forming. An intermediate was presumed to have formed that if present in high concentration would aggregate irreversibly instead of fold to the native state. Refolding of tryptophanase showed that it aggregated only with itself, suggesting specificity.
- 1981: King, at MIT, found a single amino acid folding mutant in a viral protein. Both the normal and mutant viral protein unfold at high temperature, but only the mutant would aggregate at high temperature, suggesting that aggregation could be programmed into or out of a gene
- mid 80's: The biotech industry, struggling to express growth hormone, found that a single amino acid change in bovine growth hormone completely prevented aggregation without affecting correct folding.
Protein Aggregates and Disease
This knowledge of protein folding and aggregation was soon turn toward understanding several diseases in which protein aggregates were observed which either initiated or were associated with disease. These protein aggregates were termed "amyloid deposits" and seemed to be associated and perhaps causative of several neurodegenerative diseases. The name amyloid was first used by a German pathologist, Rudolf Virchow, who in 1853 described waxy tissue deposits associated with eosinophils (a type of immune cell). These deposits seemed to resemble starch (made of amylose and amylopectin) so he termed them amyloid. All known amyloid deposits are, however, composed of protein, not starch. It now appears that these disease may be caused by improper protein folding and subsequent aggregation. Except in certain rare inherited diseases, the amyloid deposits are composed of normal proteins (often called wild-type proteins) which seems to polymerize into fibrils. Sometimes, in inherited conditions, or when mutations appear in a specific protein, the amyloid protein deposits consist of the mutant protein. The proteins in these deposited fibers are composed predominantly of beta sheets which are perpendicular to the fiber axis, while the polymerized protein usually has little beta sheet structure. Examples are given below:
- Familial amyloidotic polyneuropathy (FAP) - Affects 1/10,00 to
1/100,000 people. The protein involved is called transthyrein, which
normally exists in blood as a tetramer formed by association of 4
identical monomers. In mildly acid condition in vitro, the equilibrium
between tetramer and monomer is shifted to monomer, which can aggregate
into fibrils. This aggregation could be promoted by possible transition
to a molten globule (discussed previously with lactalbumin) like state.
This has secondary structure but loosely packed tertiary structure with
more exposed hydrophobes. If the concentration is high enough the molten
globules aggregate. In people with the disease, mutations in the protein
destabilize the tetramer, pushing the equilibrium to the monomer, which
presumably increases molten globule formation and aggregation.
Specifically, Val30Met and Leu55Pro mutations promote dissociation of
the tetramer and formation of aggregates. Conversely, Thr119Met
inhibits tetramer dissociation. The aggregates deposit in heart, lungs,
kidney, etc, leading to death.
- Light Chain Amyloidosis; Light Chain Deposition Disease - The light
chain protein is a normal component of circulating antibody molecules.
Mutants in the light chain cause a destabilization of the native state
to state similar to a molten globule, which then aggregates.
- Lysozyme amyloidosis - This protein, with extensive alpha-helix
structure, is usually involved in carbohydrate catabolism. Two mutants,
Asp67His and another, Ile56Thr (normal amino acid/number in
sequence/mutant amino acid) destabliize the protein structure (as
indicated by a decrease in the Tm of about 10 degrees C) to a
molten-globule form, which probably aggregates to fibrils characterized
by extensive beta structure.
- Alzheimers-This disease involves a defect in a protein normally found in the membrane of neurons. The protein, called beta-amyloid precursor protein (BAPP), is a transmembrane protein. A slightly truncated, soluble form is also found secreted from cells and is found in extracellular fluid (such as cerebrospinal fluid and blood). The normal function of these BAPP proteins is not yet clear. An endoprotease cleaves a small 40-42 amino acid fragment from this protein, forming the amyloid beta (Ab) protein. It is this protein or a mutant form of it which aggregates to form beta-sheet containing fibrils in Alzheimer's disease. Several mutations in different proteins have been linked to Alzheimer's, but they all seem to increase production or deposition or both of the amyloid beta protein. These deposited plaques are extracellular, and have been shown to cause neuronal damage. The are found in areas of the brain required for memory and cognition. The BAPP gene is found on chromosome 21, the same chromosome which is present in an extra copy (trisomy 21) in Downs Syndrome, whose symptoms include presenile dementia and amyloid plaques. Aggregate formation appears to be driven by increased expression of BAPP and hence amyloid beta protein. In addition, some mutants may serve to destabilize the amyloid beta protein, increasing its aggregation.
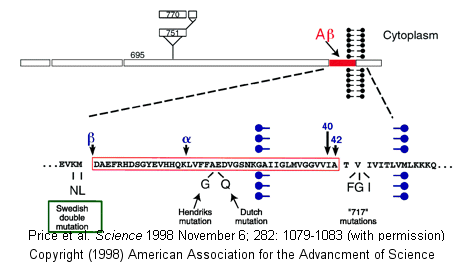
- Transmissible spongiform encephalopathies (TSEs) - Including scrapie in sheep, bovine spongiform encephalopathy (mad cow disease), and in humans Creutzfeld-Jacob Disease (CJD), Fatal Familial Insomnia (FFI),Gerstman-Straussler-Scheinker Syndrome, and Kuru (associated with cannibalism). In these fatal diseases, the brain, on autopsy, resembles a sponge with holes. In contrast to the diseases above, these diseases can be transmitted from one animal to another, but typically not between species. (However, consider the controversy with mad cow disease.) Also, the infectious agent can self-replicate in vivo. The logical conclusion is that a virus (slow-acting) is the causative agent. However, the infectious agent survives radiation, heat, chemical agents, and enzymes designed to kill viruses and their associated nucleic acids. Mathematical analyses suggested that the infectious agent in such diseases could be nothing more than a protein. Stanley B. Prusiner in the 80's isolated just such a protein which he named a prion, for proteinaceous infectious agent. Since then he and others have amassed substantial evidence to support his contention. In October 1997 he was awarded the Nobel Prize in Medicine.
H2. Prions and Disease
The normal cellular form of this protein, PrPc is highly conserved in mammals, and is widely expressed in embryogenesis. Techniques exist to delete or make ineffectual genes in mice. When a mice knockout of the PrPc (i.e. the gene for the protein was deleted in all cells) was made, the mice appeared normal. More recent data suggests however, that these mice had altered circadian rhythms and sleep patterns, which suggest a possible link to Fatal Familial Insomnia. The PrPc is a normal membrane protein in neurons. It is anchored to the membrane through a glycosyl-phosphatidyl inositol link, with the protein chain on the outside of the neuronal plasma membrane. The PrPc (without the PI link) is water soluble, protease sensitive, and consists of 42% alpha helix and 3% beta sheet.
![]() Jmol:
Updated Prion Protein, Mad Cow Disease, and Mutations
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Prion Protein, Mad Cow Disease, and Mutations
Jmol14 (Java) |
JSMol (HTML5)
The problem in the transmissible spongiform encephalopathies (TSE's) is that amyloid-like protein aggregates form which appear to be neurotoxic. The protein found in the plaques (in cases other that those that are inherited) has the same primary sequence as the PrPc but a different secondary and presumably tertiary structure. The protein found in the plaques, called the PrPsc (the scrapie form of the the normal protein) is insoluble in aqueuous solution, protease resistant, and has a high beta sheet content (43%) and lower alpha helix content (30%) than the normal version of the protein PrPc.
Figure: Cartoon Models of PrPc and PrPsc
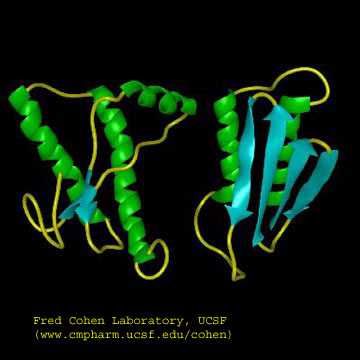
A genetic, inheritable form of disease also exists, in which a mutant form of the PrPc occurs, whose normal structure is destabilized by the mutation. The aggregates caused by the mutant form of the disease are understandable in light of the other diseases which we discussed above. The question is how does the normal PrPc form PrPsc . Evidence shows that if radiolabeled PrP*c from scrapie free cells is added to unlabeled PrPsc from scapie infected cells, the PrP*c is converted to PrP*sc! It appears that the PrPc protein has two forms not that much different in energy, one composed of mostly alpha helix and the other of beta sheet. A dimer of PrPc.PrPsc might form, which destabilizes the PrP*c causing a conformational shift to the PrPsc form, which would then aggregate. Exposure to the PrPsc form would then catalyze the conversion of normal PrPc to PrPsc . Hence, it would be transmissible by contact with just the PrPsc form of the protein. Likewise species specificity could be explained if only dimers of PrPc.PrPsc formed from proteins of the same species could occur. The inherited form of the disease would be explained since the mutant form of the normal protein would more easily form the beta structure found in the aggregate.
It has recently been found that the very same mutation in PrPc, Asp178Asn can cause two different diseases - CJD and FFI. Which disease you get depends on if you have 1 of two naturally occurring, nonharmful variants at amino acid 129 of the normal PrPc gene. If you have a Met at that position, and acquire the Asp178Asn mutation, you get CJD. If, on the other hand, you have a Val at amino acid 129 and acquire the Asp178Asn mutation, you get FFI. This disease was first observed in 1986 and has been reported in five families in the world. It occurs in the late 50's, equally in men and women. It is characterized by a progressive loss of the ability to sleep and disrupted circadian rhythms. The brain shows neuronal loss. It is known that amino acids 129 and 178 occur at the start of alpha helices, as predicted from propensity calculations. Chronic exposure to micromolar levels of synthetic fragment 106-126 of PrPc kills hippocampal neurons. This peptide also has the greatest tendency to aggregate synthetic PrPc peptides.
A series of recent studies have expanded on our knowledge of prion structure. Nelson et al. have obtained the crystal structure of a fibril aggregate made of a short peptide (7 amino acids) from the yeast prion protein Sup35. As presumably occurs in amyloid fibers, these crystals show beta-sheet structures stacked vertically to produce fibril structures. The unit of stacking appears to be pairs of beta-sheets, with the inner side amino acids of one member of the pair interacting with the inner amino acid side chains of the other member of the pair, in a process which excludes water. Similar studies by Ritter et al, using NMR and fluorescence, found pair of beta sheets to be the motif of the fibril. Using fluorescence, they identified two regions, each 15 amino acids, important in collapse to molten-globule like state for nucleation of fibril formation.
Kuru killed many members of the Fore tribe in New Guinea until the cannibalistic practice of eating dead relatives was stopped. Analysis of the genes for the prion protein in the Fore tribe and other ethnic groups in the world show two version differing by just one amino acid in all people (remember that a single gene is represented in both maternal and paternal chromosomes. That these two forms exist through the world suggest that they have been selected for by evolution and confer some biological advantage. People who have just one form of the protein are more susceptible to the development of prion diseases. Mead and Collinge have shown that about 75% of older Fore women (who had lived through cannibalistic practices) had two different prion genes, compared to about 15% of women from other ethnic groups. This high percentage suggests that these women were protected from the disease, leading through natural selection to a high percentage of heterozygotes in this defined population. The general presence of two forms of the prion gene (which probably offers protection from prion disease) suggests that cannibalism might have been widespread in our early ancestors.
There appears to be one main difference between the formation of amyloid fibers from prion proteins and others such as mutant lysozymes. If you add mutant lysozyme to normal lysozyme, the amyloid fibers contain only the mutant protein. However, if you incubate mutant prion proteins with normal prions, the normal proteins become pathological.
QED - Protein Aggregates are not just test tube artifacts, but rather matters of life and death.
![]() CDC:
Prion Disease
CDC:
Prion Disease
![]() Jsmol:
Jsmol:
![]() Protopedia
- Prions
Protopedia
- Prions
H3. Misfolding and Aggregation - Summary
Recent work has shown the proteins considered to be completely harmless can generate misfolded intermediates that aggregate to produce pre-fibril structures that are toxic to cells. This process is usually prevented in the cell by interaction of nascent forms of the proteins with chaperones, which sequester exposed hydrophobic patches and prevent aggregation. (Obviously prion proteins and the others mentioned above are exceptions). Amyloid fibers (characterized by subunits with an abnormal amount of beta-structure) can be made from many different types of proteins as noted above. Is this property specific to just a handful of proteins, or is it more common than expected from the limited examples noted so far? The new studies show that when a bacterial protein HypF is incubated at pH 5.5 in the presence of trifluoroethanol, aggregates (but not fibrils) form with enhanced beta structure. These aggregates slowly form into fibrils characteristic of amyloid protein fibers. The early aggregates (before fibril formation) proved cytotoxic. Similar results were seen with dimers and trimers (prefibril states) of the amyloid-b peptide released from cultured neurons.
A diverse group of proteins that do not share significant secondary or tertiary structure can form amyloid-like protein aggregates. Even though their monomer forms share little in common, the insoluble amyloid aggregates have a common structure in which the monomer in the aggregates has significant b-structure whose strands run perpendicular to the aggregate axis. Since it has recently been shown that almost any protein, under the "right" set of conditions can form such aggregates, the stabilizing feature of protein aggregates must be potentially found in any protein. Evidence suggests that is the presence of a polypeptide backbone, which can form stable interstrand H-bonds in beta secondary structure, and not the side-chains, that is the source of the common amyloid structure. In contrast, native, nonamyloid forms of normal proteins must arise through specific interactions of unique side chain sequence and structure, which out competes nonspecific interactions among backbone atoms found in amyloid structures. Nonspecific aggregation becomes more prevalent when buried hydrophobic side chains and buried main chain atoms become more solvent exposed. Such exposure occurs when native proteins form intermediate molten globule states when subjected to altered solvent conditions or when destabilizing mutants of the wild-type protein arise. Some mutations may alter the cooperativity of folding which would increases the fraction of nonnative proteins states. Other mutations that decrease the charge on the protein or increasing their hydrophobicity might enhance aggregation. In addition, chemical modifications to proteins (such as oxidation or deamination) might destabilize the native state, leading to the formation of the molten globule state. Once formed, this state may aggregate through sequestering exposed side chain hydrophobes or through inter-main chain H bond formation. Aggregate formation appears to proceed through the initial formation of soluble units (which may or not be more toxic to cells than the final aggregate). Aggregates are kinetically stable species. Since amyloid aggregates are cytotoxic and almost any protein can form them, albeit with different propensities, nature, through evolutionary selection, has presumably disfavored proteins with high tendencies to form such aggregates.
-
 Nature
Insights:
Protein Folding and Misfolding (2003) - Great series of articles
(PDFs).
Nature
Insights:
Protein Folding and Misfolding (2003) - Great series of articles
(PDFs).
Wasmer et al have determined a solid state NMR structure of an amyloid fiber state for a prion forming domain of the HET-s protein from the fungus P. anserina. It consists of a left-handed b solenoid.
![]() Jmol:
Jmol:
![]() Amyloid
Fibril Models from UCLA
Amyloid
Fibril Models from UCLA
Clearly accurate protein folding is required for cell viability. Aberrant protein folding clearly can be the cause of serious illness. Given the extraordinary nature of the task and its failure, the process governing protein folding must be highly regulated. The diagram below shows the steps that determine intracellular concentrations and locations of normal and aberrant protein structures.
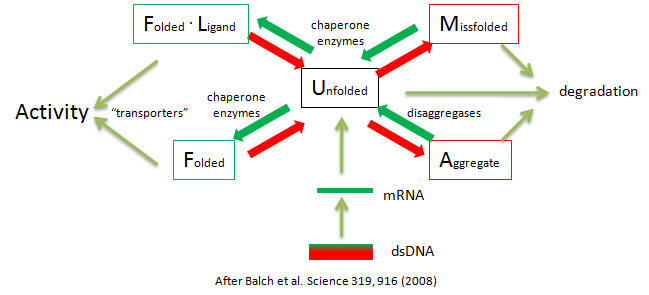
Potential therapies of diseases of proteostasis include replacing aberrant proteins, shifting the equilibria toward active forms with small ligands, or modulating the pathways with agents that influence pathways such as signal transduction, transcription, translation, degradation, and translocation using molecules like siRNAs to modulate concentrations of chaperons, disaggregases, and signal pathways.
H4. Recent Links and References
-
Wasmer, C. Amyloid Fibrils of the HET-s(218-289) Prion Form a b Solenoid with a Triangular Hydrophobic Core. Science. 319, 1523 (2008)
-
Balch, W. et al. Adapting Proteostasis for Disease Intervention. Science. 319, 916 (2008)
-
Krishnan and Linquist, Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature, 435, 765 (2005)
- Ritter et al. Correlation of structural elements and infectivity of the HET-s prion. Nature, 435, 844 (2005)
- Nelson, R. et al. Structure of the cross-b spine of amyloid-like fibrils. Nature 435, 773 (2005).
- Hardy and Selkoe. The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science 297, pg 353 (2002)
- Pepys et al. Targeted Pharmacological Depletion of Serum Amyloid P Component for Treatment of human amyloidosis. Nature. 254, 231 and 254 (2002)
- Bucciantini et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 416, pgs. 483, 507 (2002)
- Walsh et al. Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 416, pgs. 483, 535 (2002)
- Esler et al., A Portrait of Alzheimer Secretases - New Features and Familiar Faces. . Science. 293, pg 1449 (2001)
- Happarstrom et al. Trans-Suppression of Misfolding in an Amyloid Disease. Science 293 pg 2459 (2001)
- An Inflammatory Drug Prospect (for Alzheimers). Nature. 414, pg 199 (2001)
- Saborio et al. Sensitive Detection of Pathological prion protein by cyclic amplification of protein misfolding. Nature. 411, pg 810 (2001)
- Bence et al. Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science. 292, pg 1552 (2001)
- Cao et al. Elusive Protein Auditions for Several Roles (about APP in neural cells) Science. 293, pg 28, 115 (2001)
- Serio et al. Nucleated Conformational Conversion and the Replication of Conformational Information by a prion determinant. Science. 289, pg 1317 (2000)
- Chien and Weissman. the Shape of a species barrier (in prion conformation transformation). Nature. 410, pg 161. 223 (2001)
 Navigation
Navigation
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2H: Protein Aggregates and Disease

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.