Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
A: AMINO ACIDS
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 02/27/16
|
Learning Goals/Objectives for Chapter 2A: After class and this reading, students will be able to
|
A1. Amino Acid Structure
Proteins are polymers of a bifunctional monomer, the amino acid. The twenty common naturally-occurring amino acids each contain an a-carbon, an a-amino group, an a-carboxylic acid group, and an a-side chain or side group. These side chains (or R groups) may be either nonpolar, polar and uncharged, or charged, depending on the pH and pKa of the ionizable group. Two other amino acids occasionally appear in proteins. One is selenocysteine, which is found in Arachea, eubacteria, and animals. Another just recently found is pyrrolysine, found in Arachea. Shultz et al. have gone one step further. They have engineered bacterial to incorporate two new amino acids, O-methyl-tyrosine and p-aminophenylalanine. More recently, they (Chin et al.) have engineered the yeast strain Saccharomyces cerevisiae to incorporate five new unnatural amino acid (using the TAG nonsense codon and new, modified tRNA and tRNA synthetases) with keto groups that allow chemical modifications to the protein. We will concentrate only on the 20 abundant, naturally-occurring amino acids.
- Structure and Property of the Naturally-Occurring Amino Acids (Too large to include in text: print separately)
-
 Learning
Amino Acids Structure: YouTube -
Part 1 |
Part 2
Learning
Amino Acids Structure: YouTube -
Part 1 |
Part 2
Amino acids form polymers through a nucleophilic attack by the amino group of an amino acid at the electrophilic carbonyl carbon of the carboxyl group of another amino acid. The carboxyl group of the amino acid must first be activated to provide a better leaving group than OH-. (We will discuss this activation by ATP latter in the course.) The resulting link between the amino acids is an amide link which biochemists call a peptide bond. In this reaction, water is released. In a reverse reaction, the peptide bond can be cleaved by water (hydrolysis).
When two amino acids link together to form an amide link, the resulting structure is called a dipeptide. Likewise, we can have tripeptides, tetrapeptides, and other polypeptides. At some point, when the structure is long enough, it is called a protein. The average molecular weight of proteins in yeast is about 50,000 with about 450 amino acids. The large protein might be titin with molecular weight of about 3 million (about 27,0000 amino acids). A new class of very small proteins (30 or fewer amino acids and perhaps better named as polypeptides) called smORFs (small open reading frames) have recently been discovered to have significant biological activity (Science, doi:10.1126/science.1238802, 2013). These are encoded directly in the genome and are produced by the same processes that produce regular proteins (DNA transcription and RNA translation). They are not simply the result of selective cleavage of a larger protein into smaller peptide fragments.
There are many different ways to represent the structure of a polypeptide or protein. each showing differing amounts of information.
Figure: Different Representations of a Polypeptide (heptapeptide)
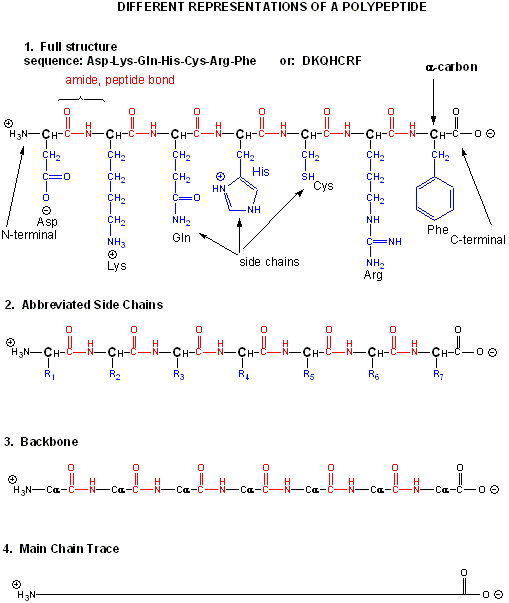
Figure: Amino Acids React to Form Proteins
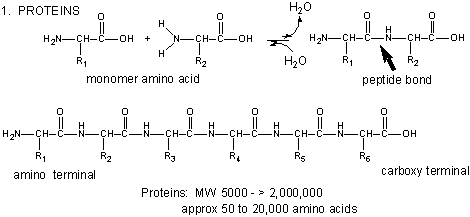
(Note: above picture represents the amino acid in an unlikely protonation state with the weak acid protonated and the weak base deprotonated for simplicity in showing removal of water on peptide bond formation and the hydrolysis reaction.) Proteins are polymers of twenty naturally occurring amino acids. In contrast, nucleic acids are polymers of just 4 different monomeric nucleotides. Both the sequence of a protein and it's total length differentiate one protein from another. Just for an octapeptide, there are over 25 billion different possible arrangement of amino acids (820). Compare this to just 65536 different oligonucleotides (4 different monomeric deoxynucleotides) of 8 monomeric units, an 8mer (84). Hence the diversity of possible proteins is enormous.
Please consult the Jmol site below dealing with amino acids. Please learn the 3 letter code for the amino acids.
![]() Jmol:
Amino
Acids
Jmol:
Amino
Acids
A2. Amino Acid Stereochemistry
The amino acids are all chiral, with the exception of glycine, whose side chain is H. As with lipids, biochemists use the L and D nomenclature. All naturally occurring proteins from all living organisms consist of L amino acids. The absolute stereochemistry is related to L-glyceraldehyde, as was the case for triacylglycerides and phospholipids. Most naturally occurring chiral amino acids are S, with the exception of cysteine. As the diagram below shows, the absolute configuration of the amino acids can be shown with the H pointed to the rear, the COOH groups pointing out to the left, the R group to the right, and the NH3 group upwards. You can remember this with the anagram CORN.
Figure: Stereochemistry of Amino Acids.
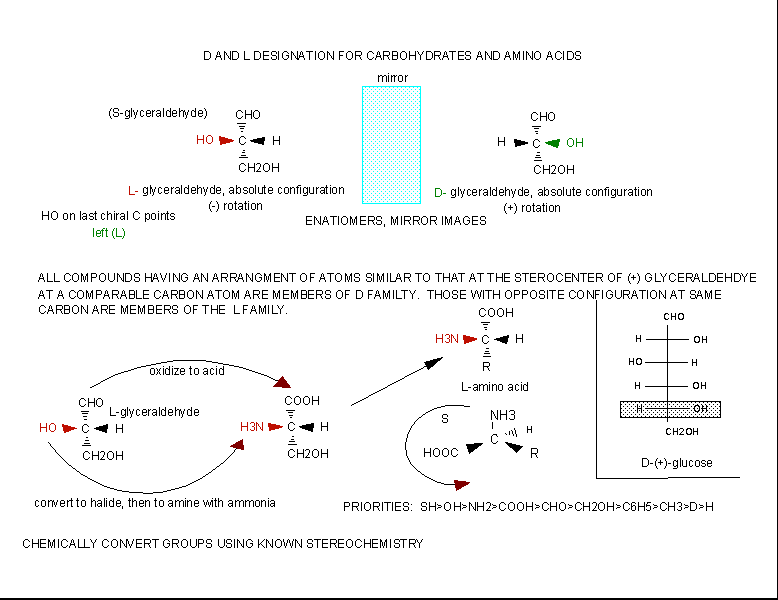
Why do Biochemistry still use D and L for sugars and amino acids? This explanation (taken from the link below) seems reasonable.
"In addition, however, chemists often need to define a configuration unambiguously in the absence of any reference compound, and for this purpose the alternative (R,S) system is ideal, as it uses priority rules to specify configurations. These rules sometimes lead to absurd results when they are applied to biochemical molecules. For example, as we have seen, all of the common amino acids are L, because they all have exactly the same structure, including the position of the R group if we just write the R group as R. However, they do not all have the same configuration in the (R,S) system: L-cysteine is also (R)-cysteine, but all the other L-amino acids are (S), but this just reflects the human decision to give a sulphur atom higher priority than a carbon atom, and does not reflect a real difference in configuration. Worse problems can sometimes arise in substitution reactions: sometimes inversion of configuration can result in no change in the (R) or (S) prefix; and sometimes retention of configuration can result in a change of prefix.
It follows that it is not just conservatism or failure to understand the
(R,S) system that causes biochemists to continue with D and L: it is just
that the DL system fulfils their needs much better. As mentioned, chemists
also use D and L when they are appropriate to their needs. The
explanation
given above of why the (R,S)
system is little used in biochemistry is thus almost the exact opposite of
reality. This system is actually the only practical way of unambiguously
representing the stereochemistry of complicated molecules with several
asymmetric centres, but it is inconvenient with regular series of molecules
like amino acids and simple sugars. "
If I told you to draw the correct stereochemistry of a molecule with 1 chiral C (S isomer for example) and I gave you the substituents, you could do so easily following the R, S priority rules. However, how would you draw the correct isomer for the L isomer of the amino acid alanine? You couldn't do it without prior knowledge of the absolute configuration of the related molecule, L glyceraldehyde, or unless you remembered the anagram CORN. This disadvantage, however, is more than made up for by the fact that different L amino acids with the same absolute stereochemistry, might be labeled R or S , which makes this nomenclature unappealing to biochemists.
- Confusion about the prefixes D- and L- : from Common Mistakes in Biochemistry Textbooks.
Monomeric amino acids have an alpha amino group and a carboxyl group, both of which may be protonated or deprotonated, and a R group, some of which may be protonated or deprotonated. When protonated, the amino group has a +1 charge, and the carboxyl group a 0 charge. When deprotonated the amino group has no charge, while the carboxyl group has a -1 charge. The R groups which can be protonated/deprotonated include Lys, Arg and His, which have a + 1 charge when protonated, and Glu and Asp (carboxylic acids), Tyr and Ser (alcohols) and Cys (thiol), which have 0 charge when protonated. Of course, when the amino acids are linked by peptide bonds (amide link), the alpha N and the carboxyl C are in an amide link, and are not charged. However, the amino group of the N -terminal amino acid and the carboxyl group of the C-terminal amino acid of a protein may be charged. The Henderson Hasselbach equation gives us a way to determine the charge state of any ionizable group knowing the pKa of the group. Write each functional group capable of being deprotonated as an acid, HA, and the deprotonated form as A. The charge of HA and A will be determined by the functional group. The Ka for the reaction is:
Ka = [H3O+][A]/[HA]. or
[H3O+] = Ka[HA]/[A].
- log [H3O+] = -log Ka + log [A]/[HA]
or pH = pKa + log [A]/[HA]
This is the (in)famous Henderson-Hasselbach (HH) equation.
The properties of a protein will be determined partly by whether the side chain functional groups, the N terminal, and the C terminal are charged or not. The HH equation tells us that this will depend on the pH and the pKa of the functional group.
- If the pH is 2 units below the pKa, the HH equation becomes, -2 = log A/HA, or .01 = A/HA. This means that the functional group will be about 99% protonated (with either 0 or +1 charge, depending of the functional group).
- If the pH is 2 units above the pKa, the HH equation becomes 2 = log A/HA, or 100 = A/HA. Therefore the functional group will be 99% deprotonated.
- If the pH = pka, the HH equation becomes 0 = log A/HA, or 1 = A/HA. Therefore the functional group will be 50% deprotonated
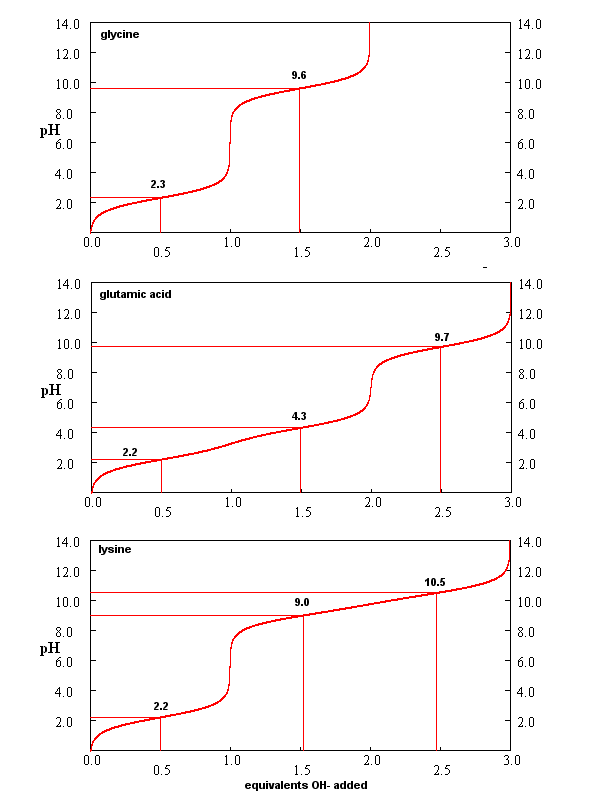
From these simple examples, we have derived the +2 rule. This rule is used to quickly determine protonation, and hence charge state, and is extremely important to know (and easy to derive). Titration curves for Gly (no ionizable) side chain, Glu (carboxlic acid side chain) and Lys (amine side chain) are shown below. You should be able to associate various sections of these curves with titration of specific ionizable groups in the amino acids.
Figure: Titration curves for Gly, Glu, and Lys
Buffer Review
The Henderson-Hasselbach equation is also useful in calculating the composition of buffer solutions. Remember that buffer solutions are composed of a weak acid and its conjugate base. Consider the equilibrium for a weak acid, like acetic acid, and its conjugate base, acetate:
CH3CO2H + H2O <==> H3O+ + CH3CO2-
If the buffer solution contains equal concentrations of acetic acid and acetate, the pH of the solution is:
or pH = pKa + log [A]/[HA] = 4.7 + log 1 = 4.7
A look at the titration curve for the carboxyl group of Gly (see above) shows that when the pH = pKa, the slope of the curve (i.e. the change in pH with addition of base or acid) is at a minimum. As a general rule of thumb, buffer solution can be made for a weak acid/base in the range of +/- 1 pH unit from the pKa of the weak acids. At the pH = pKa, the buffer solution best resists addition of either acid and base, and hence has its greatest buffering ability. The weak acid can react with added strong base to form the weak conjugate base, and the conjugate base can react with added strong acid to form the weak acid (as shown below) so pH changes on addition of strong acid and base are minimized.
-
addition of strong base produces weak conjugate base: CH3CO2H + OH- --> CH3CO2- + H2O
-
addition of strong acid produces weak acid: H3O+ + CH3CO2---> CH3CO2H + H2O
There are two simples ways to make a buffered solution. Consider an acetic acid/acetate buffer solution.
-
make equal molar solution of acetic acid and sodium acetate, and mix them, monitoring pH with a pH meter, until the desired pH is reached (+/- 1 unit from the pKa).
-
take a solution of acetic acid and add NaOH at substoichiometric amounts until the desired pH is reached (+/- 1 unit from the pKa). In this method you are forming the conjugate base, acetate, on addition of the weak base:
CH3CO2H + OH- --> CH3CO2- + H2O
-
Buffers for
pH control: Recipes based on pKas for acids, temperature, and ionic
strength
Isoelectric Point
What happens if you have many ionizable groups in a single molecule, as is the case with a polypeptide or protein. Consider a protein. At a pH of 2, all ionizable groups would be protonated, and the overall charge of the protein would be positive. (Remember, when carboxylic acid side chains are protonated, their net charge is 0.) As the pH is increased, the most acidic groups will start to deprotonate and the net charge will become less positive. At high pH, all the ionizable groups will become deprotonated in the strong base, and the overall charge of the protein will be negative. At some pH, then, the net charge will be 0. This pH is called the isoelectric point (pI). The pI can be determined by averaging the pKa values of the two groups which are closest to and straddle the pI. One of the online problems will address this in more detail
Remember that pKa is really a measure of the equilibrium constant for the reaction. And of course, you remember that DGo = -RT ln Keq. Therefore, pKa is independent of concentration, and depends only on the intrinsic stability of reactants with respect to the products. This is true only AT A GIVEN SET OF CONDITIONS, SUCH AS T, P, AND SOLVENT CONDITIONS.
Consider, for example acetic acid, which in aqueous solution has a pKa of about 4.7. It is a weak acid, which dissociates only slightly to form H+ (in water the hydronium ion, H3O+, is formed) and acetate (Ac-). These ions are moderately stable in water, but reassociate readily to form the starting product. The pKa of acetic acid in 80% ethanol is 6.87. This can be accounted for by the decrease in stability of the charged products which are less shielded from each other by the less polar ethanol. Ethanol has a lower dielectric constant than does water. The pKa increases to 10.32 in 100% ethanol, and to a whopping 130 in air!
- A great interactive web site: Amino Acid Acid/Base Titration Curves
A4. Introduction to Amino Acid Reactivity
You should be able to identify which side chains contain H bond donors and acceptors. Likewise, some are acids and bases. You should be familiar with the approximate pKa's of the side chains, and the N and C terminal groups. Three of the amino acid side chains (Trp, Tyr, and Phe) contribute significantly to the UV absorption of a protein at 280 nm. This section will dealing predominantly with the chemical reactivity of the side chains, which is important in understanding the properties of the proteins. Many of the side chains are nucleophiles. Nucleophilicity is a measure of how rapidly molecules with lone pairs of electrons can react in nucleophilic substitution reactions. It correlates with basicity, which measures the extent to which a molecule with lone pairs can react with an acid (Bronsted or Lewis). The properties of the atom which holds the lone pair are important in determining both nucleophilicity and basicity. In both cases, the atom must be willing to share its unbonded electron pair. If the atoms holding the nonbonded pair is more electronegative, it will be less likely to share its electrons, and that molecule will be a poorer nucleophile (nu:) and weaker base. Using these ideas, it should be clear that RNH2 is a better nucelophile than ROH, OH- is a better than H2O and RSH is a better than H2O. In the latter case, S is bigger and its electron cloud is more polarizable - hence it is more reactive. The important side chain nucleophiles (in order from most to least nucleophilic) are Cys (RSH, pKa 8.5-9.5), His (pKa 6-7), Lys (pKa 10.5) and Ser (ROH, pKa 13).
An understanding of the chemical reactivity of the various R group side chains of the amino acids in a protein is important since chemical reagents that react specifically with a given amino acid side chain can be used to:
- identify the presence of the amino acids in unknown proteins or
- determine if a given amino acid is critical for the structure or function of the protein. For example, if a reagent that covalently interacts with only Lys is found to inhibit the function of the protein, a lysine might be considered to be important in the catalytic activity of the protein.
Figure: A REVIEW SUMMARY OF THE CHEMISTRY OF ALDEHYDES, KETONES, AND CARBOXYLIC ACID DERIVATIVES
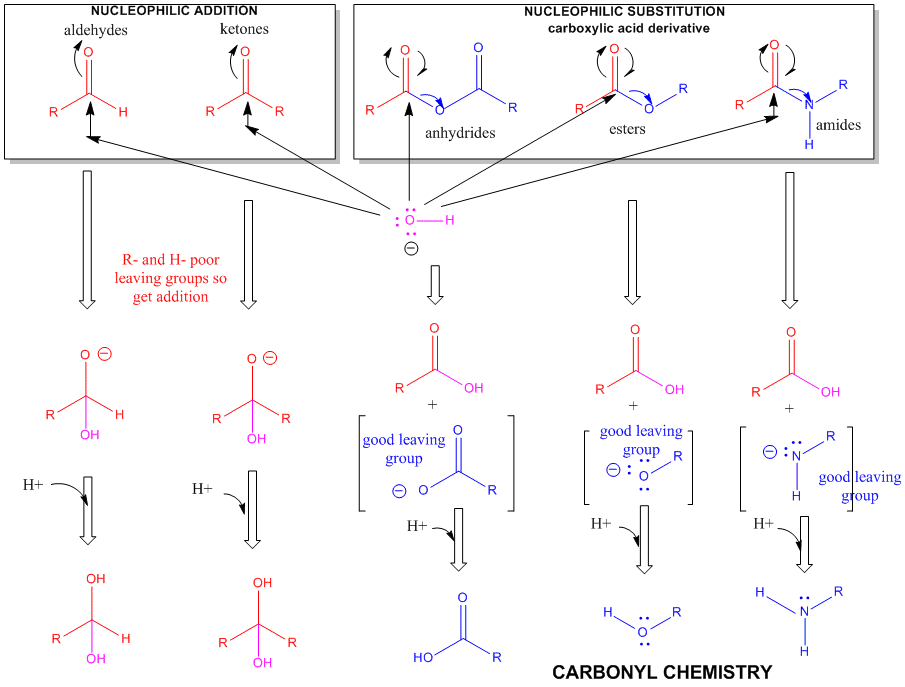
The side chain of serine is generally no more reactive than ethanol. It is a potent nucleophile in a certain class of proteins (proteases, for example) when it is deprotonated. The amino group of lysine is a potent nucleophile only when deprotonated.
A5. Reactions of Lysine
- reacts with anhydride in a nucleophilic substitution reaction (acylation).
- reacts reversibly with methylmaleic anhydride (also called citraconic anhydride) in a nucleophilic substitution reaction.
- reacts with high specificity and yield toward ethylacetimidate in a nucleophilic substitution reaction (ethylacetimidate is like ethylacetate only with a imido group replacing the carbonyl oxygen). Ethanol leaves as the amidino group forms. (has two N -i.e. din - attached to the C)
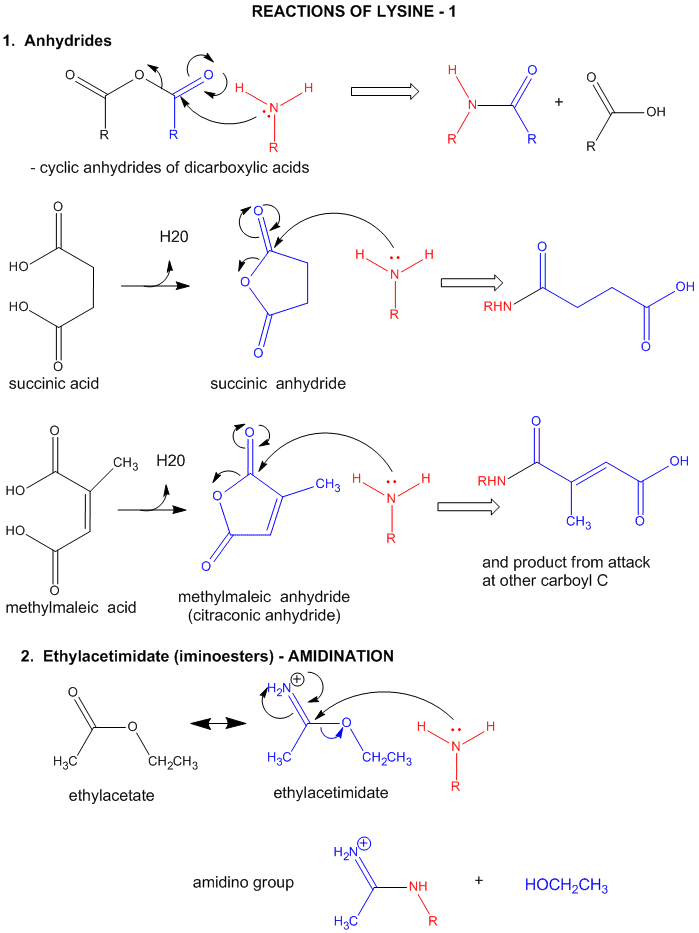
Figure: LYSINE REACTIONS 2
- reacts with O-methylisourea in a nucleophilic substitution reaction. with the expulsion of methanol to form a guanidino group (has 3 N attached to C, nidi)
- reacts with fluorodintirobenzene (FDNB or Sanger's reagent) or trinitrobenzenesulfonate (TNBS, as we saw with the reaction with phosphatidylethanolamine) in a nucleophilic aromatic substitution reaction to form 2,4-DNP-lysine or TNB-lysine.
- reacts with Dimethylaminonapthelenesulfonylchloride (Dansyl Chloride) in a nucleophilic substitution reaction.
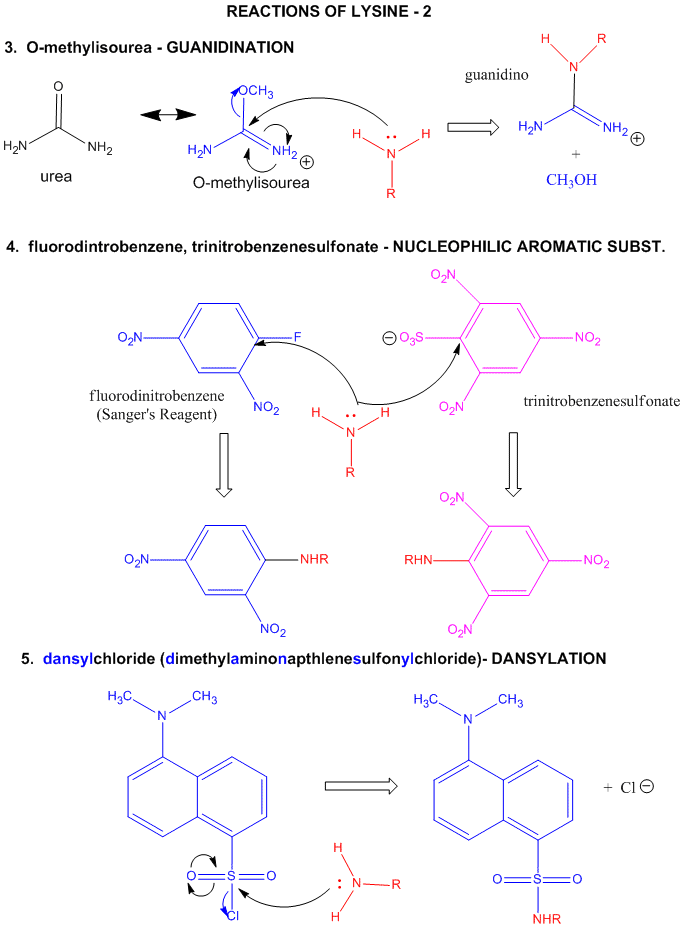
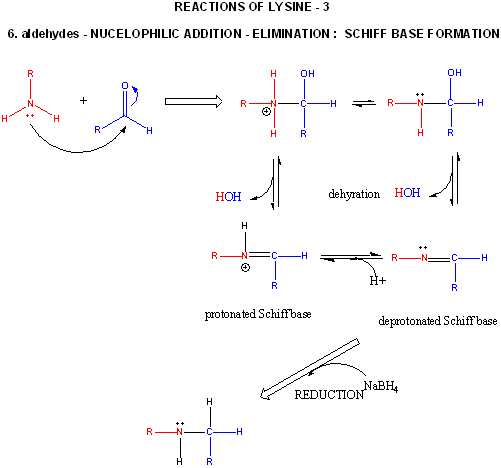
Figure: LYSINE REACTIONS 3
- reacts with high specificity toward aldehydes to form imines (Schiff bases), which can be reduced with sodium borohydride or cyanoborohydride to form a secondary amine.
A6. Reactions of Cysteine
Cysteine is a potent nucleophile, which is often linked to another Cys to form a covalent disulfide bond.
Figure: CYSTEINE REACTIONS 1
- reacts with iodoacetic acid in an SN2 rx., adding a carboxymethyl group to the S.
- reacts with iodoacetamide in an SN2 rx, adding a carboxyamidomethyl group to S.
- reacts with N-ethylmaleimide in an addition rx. to the double bond
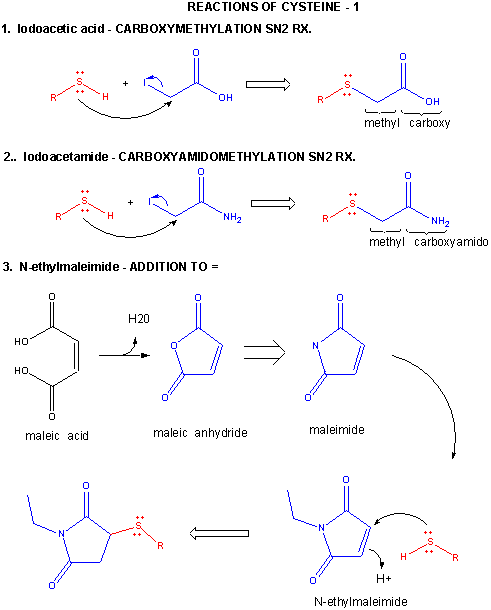
Figure: a quick review of
sulfur redox chemistry
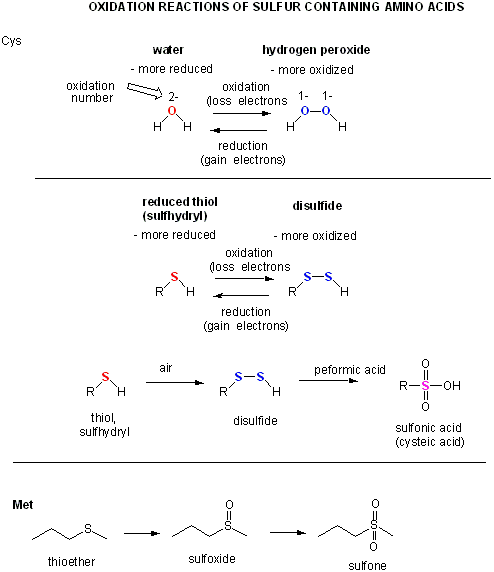
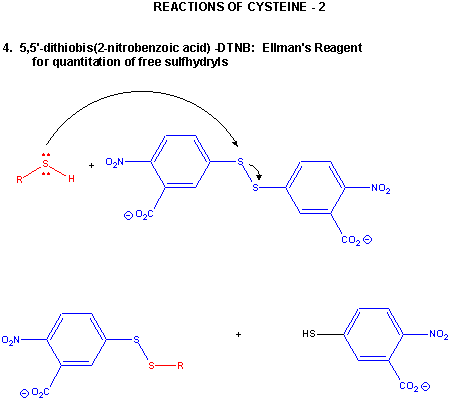
Figure: CYSTEINE REACTIONS 2
- reacts with R'-S-S-R'', a disulfide, in a disulfide interchange reaction, to form R-S-S-R'
- reacts with oxidizing agents like HCOOOH, performic acid, to form cysteic acid.
- reacts with 5,5-Dithiobis (2-nitrobenzoic acid) (DTNB or Ellman's reagent) in a RSH displacement reaction in which DTNB is cleaved and the 2-nitro-5-thiobenzoic acid anion, which absorbs at 412 nm, is released. Used to quantitate total RSH in a protein
A7. Cystine Chemistry
Two cysteine side chains can covalently interact in a protein to produce a disulfide. Just as HOOH (hydrogen peroxide) is more oxidized than HOH (O in H2O2 has oxidation number of 1- while the O in H2O has an oxidation number of 2-) , RSSR is the oxidized form (S oxidation number 1-) and RSH is the reduced form (S oxidation number 2-) of thiols. There oxidation number are analogous since O and S are both in Group 6 of the periodic table and both are more electronegative than C.
A quick review of redox reactions and oxidation numbers.
Figure: DISULFIDE - CYSTINE - REACTIONS
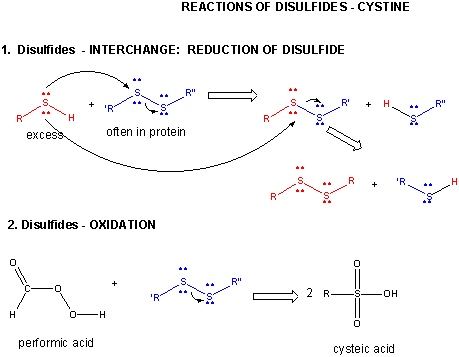
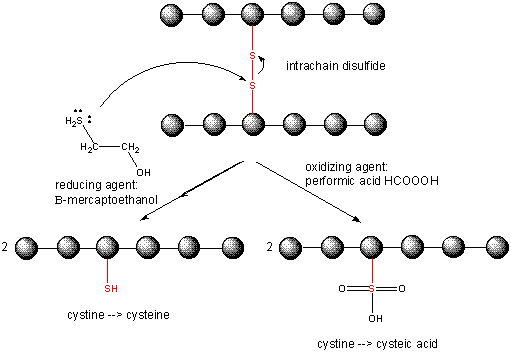
When a protein folds, two Cys side chains might approach each other, and form an intrachain disulfide bond. Likewise, two Cys side chains on separate proteins might approach each other and form an interchain disulfide. Such disulfides must be cleaved, and the chains separated before analyzing the sequence of the protein. The disulfide in protein can be cleaved by reducing agents such as beta-mercaptoethanol, dithiothreitol, tris (2-carboxyethyl) phosphine (TCEP) or oxidizing agents which further oxidizes the disulfide to separate cysteic acids.
Figure: Disulfide Oxidizing Agents - b-mercaptoethanol, dithiothreitol, and phosphines
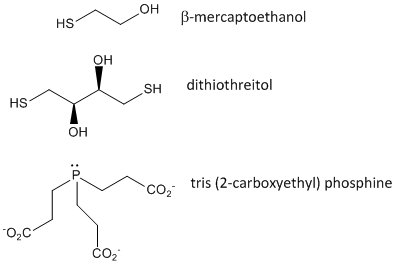
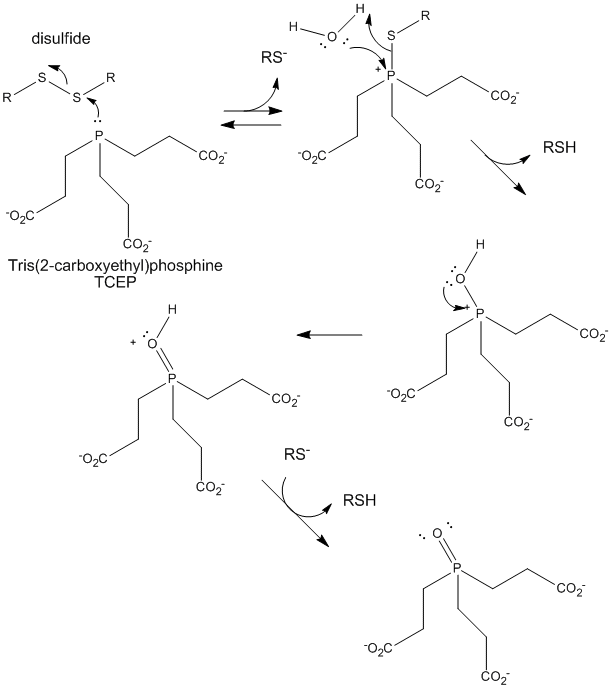
Figure: TCEP reduction of disulfides
The inside of cells are maintained in a reduced environment by the presence of many "reducing" agents, such as the tripeptide g-glu-cys-gly (glutathione). Hence intracellular proteins usually do not contain disulfides, which are abundant in extracellular proteins (such as those found in blood) or in certain organelles such as the endoplasmic reticulum and mitochondrial intermembrane space where disulfidesc can be introduced.
Figure:
Cleaving Disulfide Bonds in Proteins
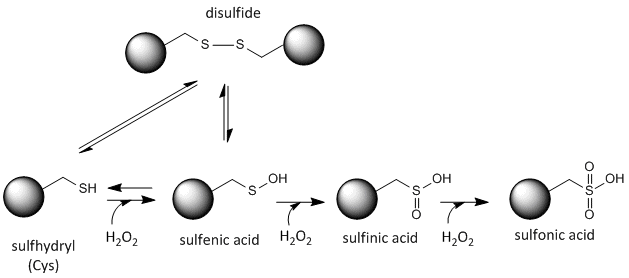
Cysteine Redox Chemistry
The sulfur in cysteine is redox-active and hence can exist in a wide variety of states, depending on the local redox environment and the presence of oxidzing and reducing agents. A potent oxidizing agent that can be made in cells is hydrogen peroxide, which can lead to more drastic and irreversible chemical modifications to the Cys side chains. If a reactive Cys is important to protein function, then the function of the protein can be modulated (sometimes reversibly, sometimes irreversibly) with various oxidizing agents, as shown in the figure below.
Figure: Redox state of Cysteine
A8. Reactions of Histidine
Histidine is one of the strongest bases at physiological pH's. The nitrogen atom in a secondary amine might be expected to be a stronger nucleophile than a primary amine through electron release to that N in a secondary amine. Opposing this effect is the steric hindrance by the two attached Cs of the N on attach on an electrophile . However, in His, this steric effect is minimized since the 2Cs are restrained by the ring. With a pKa of about 6.5, this amino acid is one of the strongest available bases at physiological pH (7.0). Hence, it can often cross-react with many of the reagents used to modify Lys side chains. His reacts with reasonably high selectivity with diethyl pyrocarbonate.
Figure: REACTIONS OF HISTIDINE
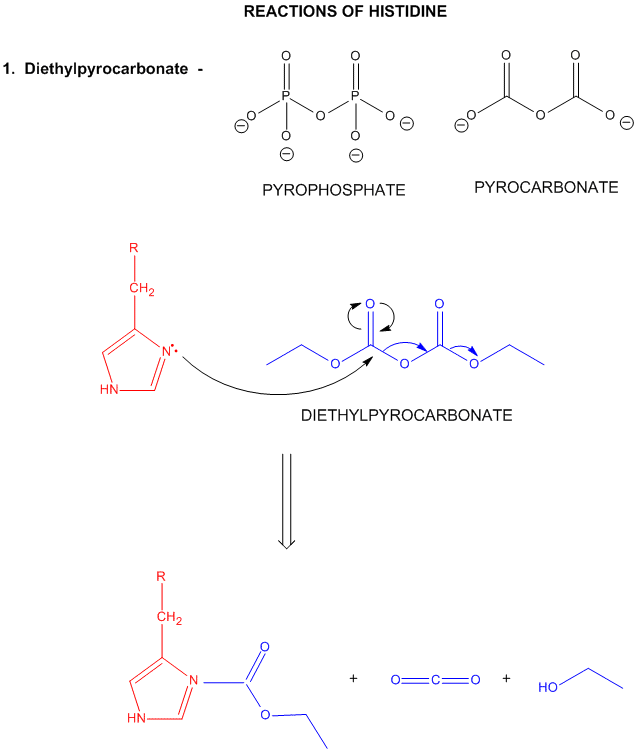
Figure: Where is the H on His? Where is the Charge?
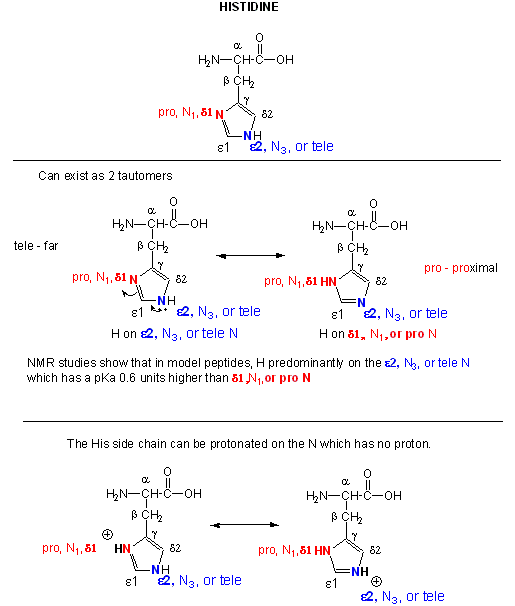
A9. In vivo Post Translational Modification of Amino Acids
Amino acids in naturally occurring proteins are also subjected to chemical modification within cells. These modifications alter the properties of the amino acid that is modified, which can alter the structure and function of the protein. Most chemical modifications made to proteins within cells occur after the protein is synthesized in a process called translation. The resulting chemical changes are termed post-translational modifications.
Figure: Post-translational modification of proteins
![]()
Here is a list of post-translational modification from the Swiss Institute of Bioinformatics:
-
PDOC00001 1 N-glycosylation site
-
PDOC00004 1 cAMP- and cGMP-dependent protein kinase phosphorylation site
-
PDOC00005 1 Protein kinase C phosphorylation site
-
PDOC00006 1 Casein kinase II phosphorylation site
-
PDOC00007 1 Tyrosine kinase phosphorylation site
-
PDOC00008 1 N-myristoylation site
-
PDOC00009 1 Amidation site
-
PDOC00010 1 Aspartic acid and asparagine hydroxylation site
-
PDOC00012 1 Phosphopantetheine attachment site
-
PDOC00013 1 Prokaryotic membrane lipoprotein lipid attachment site
-
PDOC00342 1 Prokaryotic N-terminal methylation site
-
PDOC00266 1 Prenyl group binding site (CAAX box)
-
PDOC00687 2 Intein N- and C-terminal splicing motif profiles
A10. General Links and References
- The Amino Acid Repository from the Image Library of Biological Molecules. Contains a great data base of properties about amino acids.
- Amino Acid Information form Rockefeller University.
References
- Chin, J. et al. An expanded eukaryotic genetic code. Science. 301. pg 964 (2003)
- Schultz, P. et al. JACS, last week January, 2003
- A New Twist on Chirality (how homochirality in amino acids may have developed). Science. 292, pg 2021 (2001)

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.