| |
|
|
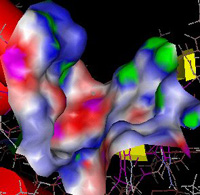
Low Molecular Weight Protein Tyrosine Phosphatase (LMW PTP)
Enzymes that cleave phosphate groups from proteins are
abundant in all cells. These enzymes, called phosphatases, regulate the
activity of phosphorylated proteins, rendering the dephosphorylated proteins
either active or inactive. Phosphatases are key proteins in the regulation
of cells growth and metabolic state. I am studying human low molecular protein
tyroysl phosphatase (PTP). Dr. McIntee (Chemistry Dept) and I are
collaborating to study the natural protein substrates of this enzyme
and to develop drugs to inhibit its activity.
The protein is made from a cDNA inserted into a plasmid,
PGEX-6P1 which is used to transform E.
Coli. Gene expression is induced by addition of IPTG, which activated the
lac promoter, leading to the formation of a GST-PTP fusion protein. The active site of the enzyme, where the chemical cleavage of the
phosphate group occurs, must bind phosphate groups which are covalently
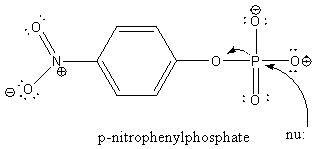
attached to target proteins. The activity of the enzyme can be monitored easily in
solution using p-nitrophenol phosphate which is cleaved by the enzyme to produce
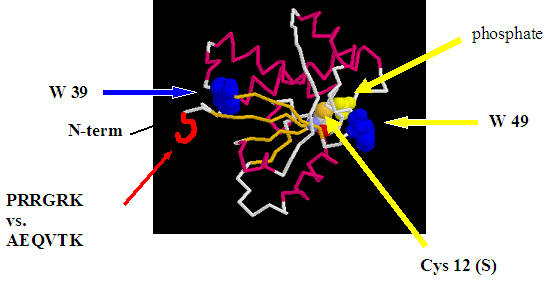
a yellow solution. An active site cysteine (Cys 12) acts as a
nucleophile in the cleavage reaction. The enzyme binds phosphate in the
active site, which competitively inhibits the enzyme.
My research group has made a series of mutations in the DNA for
PTP
and we are presently studying the effect of these mutants on its activity. The mutants can be divided into two groups:
1.
Active site mutant: The active site Cys 12 (C12) is replaced for a Ser (C12S). This makes the enzyme catalytically inactive, unable to cleave
phosphate groups from proteins. However, this mutant will still be able to bind phospho-proteins. We will use this mutant in future research to bind to natural
phospho-proteins in epithelial and fat cells, and to identify the binding sites
on those phospho-proteins for PTP.
2.
Nonactive site mutant:
PTP
contains two tryptophans (W). We have made two different mutants, changing
a single W to phenyalanine (F) in each one, thereby producing two mutants that
contain only one W residue. We have changed W49,
an amino acid that is near the active site and which fluoresces, to F
which does not fluoresce. We have also made a second mutant to change W39, located
on the opposite side from the active site, to a phenylalanine. Fluorescence
from the mutant with only a single W at position 49 will be sensitive to the
environment of the active site, while the other mutant, containing a single W at
position 39, will be used to detect changes in protein structure away from the
active site and serve as a control.
The activity of the two mutant proteins, as measured by cleavage of
p-nitrophenyl phosphate and fluorescence, in the presence and absence of
different inhibitors of the enzyme, will be used to better understand how the
structure of the enzyme influences its activity. In addition, the stability and
unfolding of the protein will be studied used fluorescence from the single
tryptophan-containing mutants.
We have also made a double mutant: C12S/W39F,
producing a protein that can not cleave phosphates from target proteins (C12S)
and which has a single W at position 49 (W39F) which will be used to monitor phospho-protein binding to the double mutant by monitoring fluorescence changes
in W49.
Additionally, we are studying the protein and its
interaction with inhibitors through in silico computer modeling of the
protein uisng VMD/NAMD and Autodock.
Applications of Fluorescence Measurements in Biochemistry
My second project involves using the spectrofluorometer to
study a range of biological questions. When molecules absorb UV or visible
light, electrons are excited to higher energy levels. The excited electrons can
�relax� back to lower energy levels by losing energy through collisions or by
emitting photons of lower energy than the original excitation photons. This
emission is called fluorescence. In contrast to simple absorbance properties,
fluorescence emission is extremely sensitive to the environment of the
flourophore, the molecule that emits. The properties of biological molecules
can be studied using fluorescence. Two types of fluorophores are used.
Intrinsic fluorophores are part of the actual molecule (for example a tryptophan
side chain in a protein). Extrinsic fluorophores (like fluorescein) can be
attached (covalently or noncovalently) to proteins, lipid aggregates and DNA.
We develop research projects using fluorescence to
study the structural transition and binding interactions of proteins, lipids,
and DNA.
My Research Group
In the summer of 2008 I have two research students, Rob
Hvalicek (SJU) and Yang Lin from Southwest University (SWU), Beibei, Chongqing,
Peoples' Republic of China.
Here's a list of some of my past research students and a
bit about what they are presently doing;
| Year |
Student, Degrees| Presently ... (last known position/status
as of 7/16/08) |
| 08-09 |
Robert Hlavacek, Senior, SJU Yang Lin,
Senior, SWU |
| 07-08 |
Andrew Hipp, applying to medical school
Jing Ying (SWU) |
| 06-07 |
Nick Menthe, M.S. xxx, | Presently Adjunct Faculty, Biology
Department, CSB/SJU Wang Jing (SWU) |
| 05-06 |
Claire Hoolihan, MPH, Environmental Health,
University of Minnesota | Presently ... Nick Briese, medical
student, University of Minnesota |
| 03-04 |
Adam Barker, medical student ? | |
| 02-03 |
Dr. Dustin Lorentz, M.D., University Minnesota |
Presently ... |
| 01-02 |
Elizabeth Cody,
Dr. Jennifer Klein, Ph.D. Biochemistry, University of
Minnesota | Presently ...
Dr. Anna Selmecki, Ph.D.,Molecular, Cellular,
Developmental Biology & Genetics, University of
Minnesota | Presently ... |
| 00-01 |
Dr. Jason Bartos, Ph.D. Pharmacology, University of
Iowa. | Presently medical student, Stanford University
Dr. Karla Ziegelmann-Fjeld, Ph.D Biochemistry, Michigan
State University, | Presently at
MicroBioLogics
Dr. Aaron Krych, M.D., Mayo Medical School, | Presently
Resident in Department Orthopedic Surgery, Mayo Clinic |
| 99-00 |
Jessie Odenthal | Presently ..., |
| 98-99 |
Dr. Amanda Sinness, M.D. North Dakota State
University | Presently ... Katie Garvey (nutrition)
Presently ... |
| 97-98 |
Eric Schneider |Presently ...
Ryan Rubischko | Presently ... |
| 96-97 |
Dr. Scott Loecken, M.D. University of Minnesota |
Presently ... |
| 95-96 |
Dr. Michael Kennedy,
Ph.D., Mayo Graduate School, | Presently Research Assistant
Professor and Director, Educational and Research Programs,
Northwestern University |
| 94-95 |
Jill Funk | Presently ... Dr. Jill Holbrook,
M.D./Ph.D University Wisconsin, Madison. | Presently
Post-doc in Molecular Medicine
Partnership Unit, European Molecular Biology Laboratory, Department for
Pediatric Oncology, Hematology and Immunology, University Hospital
Heidelberg |
| 93-94 |
Dr. Manu Chakravarthy,
M.D/Ph.D., University of Texas Medical School and the M.D.
Anderson Cancer Center; | Presently Instructor of Medicine,
Washington University
Dr. Rob Bellin,
Ph.D. Biochemistry, Iowa State University | Presently Associate Professor
Biology, Holy Cross
Mary Pennas | Presently ... |
| 92-93 |
Jeffrey Vos | Presently ... |
| 91-92 |
Dr. Steve Van der Louw, Ph.D. Chemistry, Iowa State University |
Presently ... Dr. Brenda Weyer (Sorensen),
Ph.D. Biochemistry, University of Iowa. | Presently in Biochemistry
Department, University of Iowa. |
Grants-Net:
Supported by HHMI and AAAS
11/24/2015
Return to My Home Page
|