Reactivity in Chemistry
Aliphatic Nucleophilic Substitution
NS9. Enolate Nucleophiles
Enolates and related nucleophiles deserve a closer look because they are very common and because they have their own issues of regiochemistry.
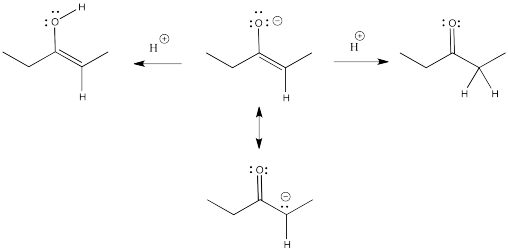
Remember, an enolate is just the conjugate base of an enol. An enolate can also be thought of as the conjugate base of a related carbonyl. Because the enolate is a delocalized anion, it can be protonated in two different places to get two different conjugates.
Figure NS9.1. The relationship between a ketone, an enol, and an enolate anion.
Enols typically are not seen because of a rapid equilibrium with that related carbonyl compound. As soon as an enol forms, if there is any way for it to transfer a proton to get to the carbonyl, it will do so. This kind of equilibrium is called "tautomerism", involving the transfer of one proton from one place to another within the molecule. The enol and its related carbonyl are referred to as "tautomers". "Tautomers" describes the relationship between these two molecules.

Figure NS9.2. Ketones and enols are tautomers of each other
Tautomerism is the relationship between isomers that differ by the position of one proton. Enamines are very similar to enolates, but with a nitrogen atom in place of the oxygen. Hence, they are amines instead of alcohols.

Figure NS9.3. Enamines are tautomers of imines.
Enamine, enolates and enols are all turbo-charged nucleophiles. The nucleophilic atom is the alpha carbon. Although that carbon can be thought of as a double bonded carbon, with no lone pair, that position is motivated to donate electrons because of pi donation from the oxygen (or nitrogen).
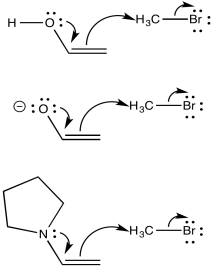
Figure NS9.4. Enols, enolates, and enamines are good nucleophiles.
One of the complications that can arise with these nucleophiles has to do with asymmetry about the carbonyl (or the would-be carbonyl). If one alpha position next to the carbonyl isn't the same as the other one, two possible enolates could result from removal of a proton.
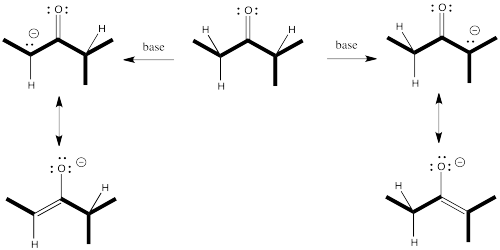
Figure NS9.5. An asymmetric ketone may lead to two different enolates.
That means we potentially have two different nucleophiles from the same
starting compound. Sometimes, mixtures of products result from enolate
reactions.
Nevertheless, enolates and enamines are very broadly used in the synthesis of important things like pharmaceuticals, precisely because they can be controlled so well. If you want the enolate on one side of the carbonyl -- we'll call it the more-substituted side -- then you can have it. If you want the enolate on the other side of the carbonyl -- the less-substituted side -- you can have that, instead.
To get the proton off and turn a carbonyl compound into an enolate requires a base. Some control over which proton is removed might come from the choice of base. Let's think about what is different about those two sides of the carbonyl. One side is more substituted. It has more stuff on it. It's more crowded. Maybe to get the proton off the more crowded position, you need a smaller base.
Conversely, to get the proton exclusively from the least crowded position, and have very little chance of getting it from the more crowded spot, you could use a really big base.
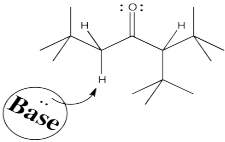
Figure NS9.6. Steric crowding can play a role in which enolate is formed.
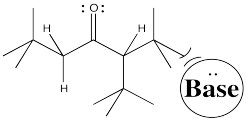
Figure NS9.7. A bulky base may be unable to approach the more crowded α proton.
So let's go all the way through a reaction, looking at two possible nucleophilic substitution products. Suppose we take 2-methylpentan-3-one, deprotonate it to make an enolate anion, and add chloroethane to complete the reaction. The overall reaction is called an α-alkylation, because the alkyl electrophile that is added goes to the alpha position, next to the carbonyl.
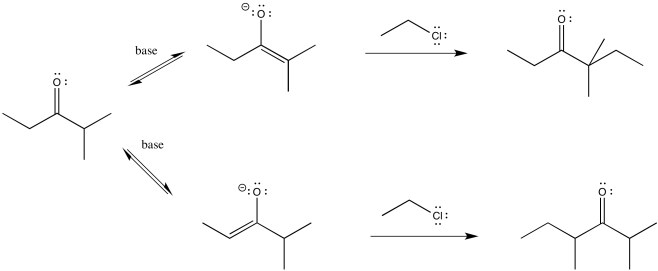
Figure NS9.8. Two different enolates leading to two different substitution products.
You can see the two different products of the reactions: one of them is 4,4-dimethylhexan-3-one. The other one is 2,4-dimethylhexan-3-one. From what we have said so far, if we use a big base, maybe it won't fit as well on the more hindered side (the top pathway above), so it will go to the least-hindered side (the bottom path).
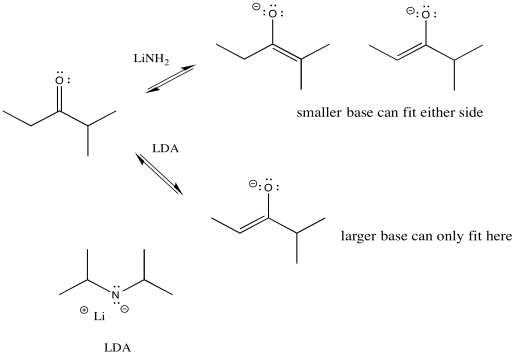
Figure NS9.9. LDA is a bulky base that takes the proton from the less crowded side.
We can make the base bulkier by adding alkyl substituents. If it just has little hydrogens attached to the basic atom, it won't be very crowded. Also, in this case we used amide bases because they are very strong bases. (Amide is the inorganic chemistry term for the conjugate base of ammonia, or NH2-.) There is an equilibrium between a ketone and an enolate. A stronger base pushes the equilibrium all the way to the enolate side, with no leftover ketone.
So if we need to take the lower pathway, we have found a way to do it. LDA (lithium diisopropylamide) will take the least hindered proton and alkylation will occur on that side. The trouble is, the much smaller lithium amide can fit on either side. It has little preference under these conditions, so it leads to a mixture of products.
How can we force things through the upper pathway?
There's something else about enolates that is apparent only when you look at the ions in one resonance form. Enolate ions can be thought of as alkenes, of course. Depending on which proton we remove, we get two different alkenes. There may be factors that make one of these two alkenes more stable. If so, there may be ways to form that one instead of the other.
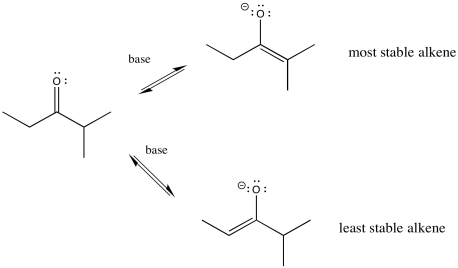
Figure NS9.10. The more-substituted enolate is more stable.
In general, more-substituted alkenes are more stable than less-substituted ones. The more substituted alkene is formed via loss of the proton at the more crowded position. Enolate stability should influence things, and it does, but there are some subtleties involved. To understand the subtleties, we are going to need to look at the potential energy diagram for enolate formation.
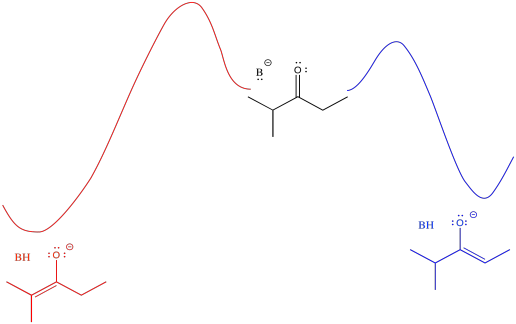
Figure NS9.11. There is a higher barrier to form the more-stable enolate than the less-stable one.
The barrier might be a little higher going from the middle of the picture to the left than going from middle to right (it's more crowded, after all). However, once the reaction gets over that barrier, it settles out in a more stable place (the enolate is more stable). An initially greater investment, in this case, yields a greater return in the end.
An alternative view of that surface places the reactant on the left and both products on the right. The two possible pathways are then superimposed on each other. The conclusion we draw is the same: the barrier is higher along the red path than the blue path because of steric hindrance. Eventually, the red path leads to the lower energy product.
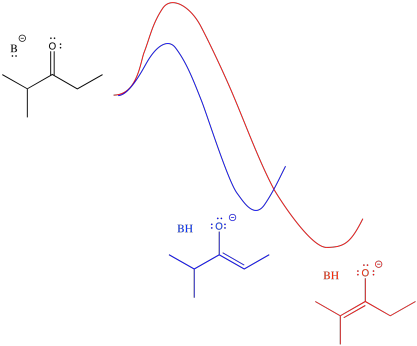
Figure NS9.12. Superimposed reaction profiles for formation of the two enolates.
Just looking at the diagram, we can infer that it takes more energy to get over the barrier leading to the more stable enolate. If we supply more energy to start with, we might have a better chance of reaching the more stable enolate. Of course, the easiest way to do that is to heat the reaction up. If we don't want to form the more stable enolate, we could cool the reaction down. That way, there wouldn't be enough energy to get to the more stable enolate.
Forming a product based on its relative stability means relying on thermodynamics. Getting to the thermodynamic product means you have to give the system a chance to find its way there. One way to do that is to allow the deprotonation to happen reversibly. Given multiple chances, the more stable enolate will form eventually.
An obvious way to allow reversibility is to conduct the reaction in a protic solvent, such as ethanol. If the enolate takes a proton from ethanol, it becomes a ketone again. It has a second chance to get deprotonated again. Eventually, it should form the most stable enolate.

Figure NS9.13. Enolate formation is reversible in the presence of alcohols.
On the other hand, if you intend to take the proton off the least substituted position, you don't want any reversibility. Given the chance, the wrong enolate will eventually form. You want to just take what you get the first time, which is more likely to be the blue pathway because of its lower barrier. Earlier, we used a bulky amide base to form the least stable enolate because we didn't want to allow the possibility of second chances.

Figure NS9.14. Enolate formation is not reversible in the presence of secondary amines.
This is a case in which we need kinetic control to get one product: we want the least-substituted enolate, and we depend on it forming more quickly than the ther enolate. After all, if the other enolate forms, it is more stable; it isn't likely to come back.
There are some even more subtle factors that are used to help things along just a little bit more. For example, the counterions for the bases can have a modest effect one way or the other. The effect has to do with the degree of covalency between the counterion and the enolate. For example, lithium is a smaller, harder alkali ion that forms strong bonds with oxygen. Potassium is a larger, softer alkali ion that forms weaker bonds with oxygen. We are just using these terms in a relative way; lithium and potassium ion are both hard, but lithium ion is even harder than potassium ion.
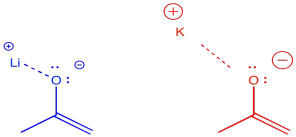
Figure NS9.15. Different alkali cations have different degrees of covalency with oxygen anions.
If the enolate is bound to its counterion, it is less likely to undergo reversal to the ketone. Lithium counterions are sometimes used to promote formation of the kinetic enolate. Potassium counterions are sometimes used to promote formation of the thermodynamic enolate.
There is one final factor to talk about, which comes up in the case of sodium hydride. The use of sodium hydride generally leads to the thermodynamic enolate. That may be a surprise. After all, sodium hydride is a very strong base. We would not expect the counterion, H2, to be acidic. That means the use of sodium hydride allows only one chance to get to the thermodynamic product. So how does NaH manage to do that?
The answer appears to be related to phase. NaH is an insoluble solid. As a result, the reaction between NaH in the solid phase and the ketone in the solution phase happens only at the surface of the NaH. This reaction is very slow. It happens over a long period of time. Remember, time was one of the factors that favoured formation of the thermodynamic product.
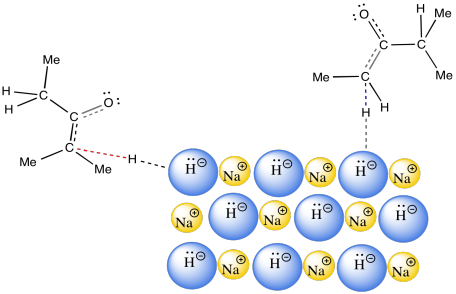
Figure NS9.16. Sodium hydride is an insoluble, slow base.
A long reaction time allows for equilibration between possible enolate ions. The thermodynamically more stable enolate should dominate this equilibrium. The trouble is, the pathway that establishes the equilibrium between the possible enolate ions requires proton transfers. As a very strong base, NaH is never used in the presence of protic solvents or acid. If it were used in a protic solvent such as water, NaH would just decompose to H2 gas and NaOH.
So what happens if NaH takes the less hindered proton, forming the less stable enolate? How does it eventually form the more stable enolate? The proton source in this case is other ketones. Because the deprotonation is so slow, there are plenty of remaining ketone molecules throughout most of the course of the reaction. They can donate protons to less stable enolates, eventually forming the more stable enolate.
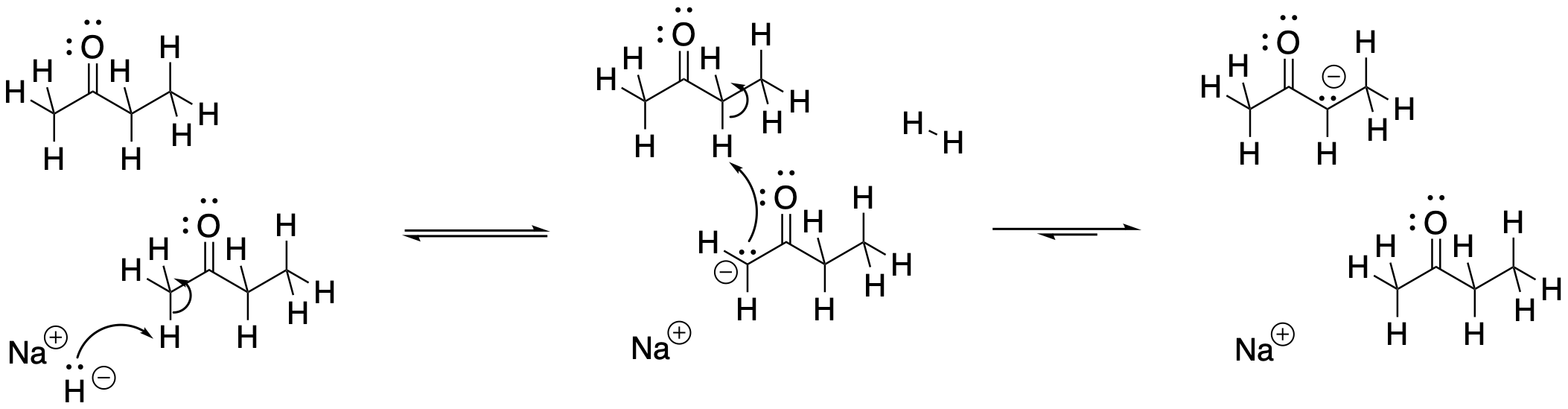
Figure NS9.17. Slow proton transfer converts one enolate into another.
Problem NS9.1.
Fill in the blanks in the following synthesis. Includes: aliphatic nucleophilic substitution, silyl ethers, carboxylic substitution, carbonyl addition (anionic nucleophiles, neutral nucleophiles, enolates)
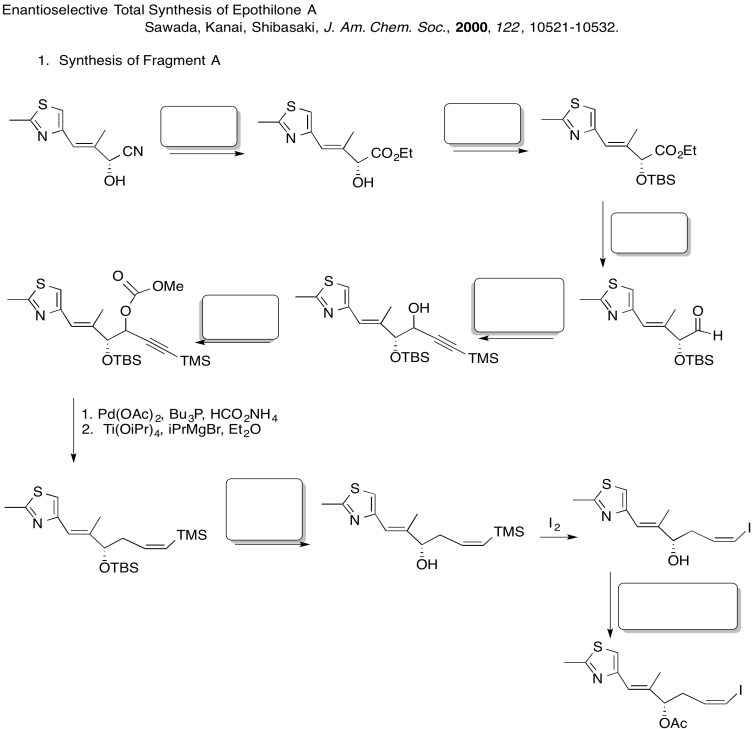
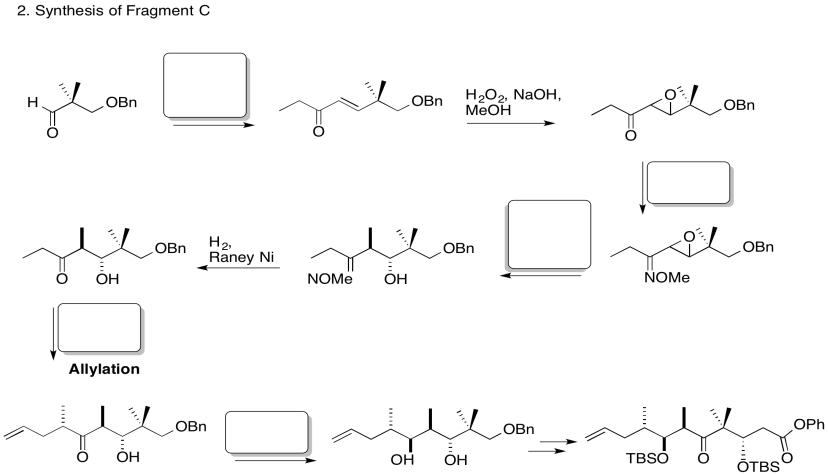
3. Stereocontrol of the allylation step:
Is the base used a kinetic or a thermodynamic base? Why?
Explain the stereocontrol of the allylation step in the preparation of Fragment C (shown here). Be sure to include pictures.
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's University (retired) with other authors as noted). It is freely available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by Chris Schaller is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Navigation: