Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 5 - BINDING
C: MODEL BINDING SYSTEMS
BIOCHEMISTRY - DR. JAKUBOWSKI
3/29/16
|
Learning Goals/Objectives for Chapter 5C: After class and this reading, students will be able to
|
C6. Conformational Selection
In our study of hemoglobin structure in the MWC model, we developed the idea that there were two forms of hemoglobin in solution, the taut and relaxed form, which are pre-exisiting and interconvertible even in the absence of dioxygen. Oxygen was presumed to bind preferentially to the relaxed form. In the KNF model we saw that ligand binding can induce conformational changes in adjacent subunits, promoting cooperative binding of ligand. In general these two models distill down to combinations of two simpler models. The first might be called the conformational selection in which ligand binds tightly to a preexisting conformations in a "lock and key manner" without inducing subsequent macromolecular conformational change. Alternatively, the ligand might bind loosely and then alter the macromolecular conformation to produce tighter binding, an example of an induced fit model. For the binding of dioxygen to hemoglobin, thermodynamic cycles could be drawn showing either binding of ligand and subsequent conformational changes in protein structure or conformational changes in protein structure proceeding binding. Is there additional evidence to support the conformational selection model of binding of ligand to a protein that can, in the absence of ligand, exist in two conformations? The answer is yes.
Antibodies are immune system protein molecules than can bind "foreign" molecules and target them for biological neutralization. Many crystal structures have been determined of antibodies in the presence or absence of a "foreign" ligand molecule. In these cases, the conformation of the bound antibody is different from that of the free. Either an induced fit model for ligand binding or a lock and key model of binding of ligand to one of two different pre-existing conformations of the antibody could account for this observation. These different mechanisms could be differentiated experimentally by stop-flow kinetic technique since both display slow and fast phases that are affected differently by ligand concentration. Theoretically, in the induced fit model, only one ligand type could bind to the antibody which would undergo a conformational rearrangement to produce tighter binding. However, in the two preexisting conformational models, a different structural ligand might bind to each of the two main antibody conformations. James et al. have recently shown through stop flow kinetics techniques (to investigate binding) and x-ray crystallography (to investigate final structures) that one antibody molecule can, through existing in two different preexisting conformations, bind two different ligands (antigens). One antibody conformations binds small aromatic molecules with low affinity (including the small molecule 2,4-dinitrophenol, the immunizing molecule or hapten) and then rearranges to produce a high affinity binding complex in which the DNP is bound in a narrow cavity (reducing the effective off rate of the bound ligand. A second antibody conformation binds a protein ligand over a broad, flat binding site of the antibody molecule.
Lange (2008) et al, using a NMR technique, residual dipolar coupling, that allows sampling of structures in the microsecond time scale, have shown that the solution structure of ubiquitin (which we modeled in our first lab), in the absence of ligand, exists in an ensemble of conformational states. More importantly, these different conformational states are identical to those found in the 46 crystal structure of ligand complexed to various protein ligands, strongly supporting the concept of conformational selection. In all likelihood, a combination of both induced fit and conformational selection probably occurs within a 3D energy landscape in which an initial binding encounter by either a lock and key fit to the "optimal fit" conformer or to a higher energy conformer in which the bound state relaxes to a lower energy through the induction of shape changes in the binding protein.
Figure: Conformational Selection vs Induced Fit Binding (after Boehr and Wright, Science 320, 1429 (2008)
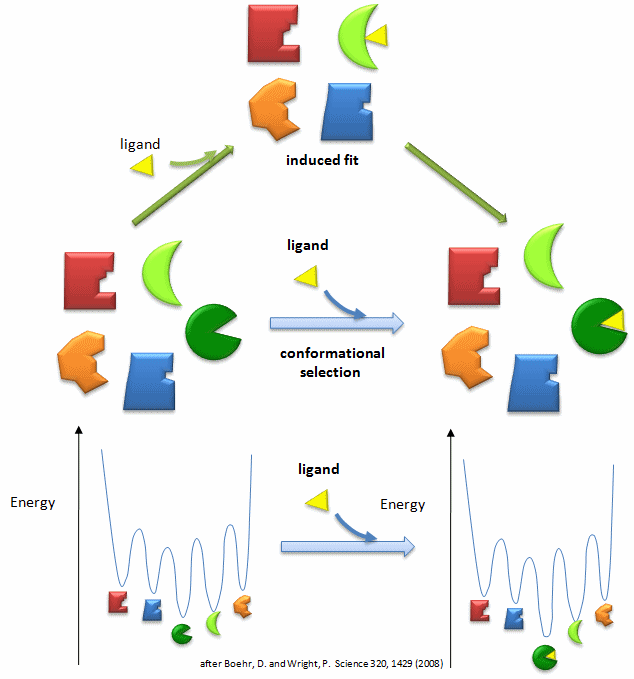
An interesting experimental model to distinguish conformational selection versus induced ligand binding was offered by Rea et al. They studied rabbit ileal bile acid binding protein (I-BABP). The wild type protein has a helix-turn-helix motif at its N terminus. They produced a mutant (Δa-I-BABP) that replaced this motif with a Gly-Gly-Ser-Gly linker, causing the protein to unfold. Next they conducted binding and folding studies on addition of taurochenodeoxycholate (TCDC) using stopped-flow fluorescence to measure the binding behavior. They wished to distinguish between two distinct mechanisms – folding before binding (or conformational selection) and binding before folding (or induced-fit model). The data support a two phase model. One phase did not depend on ligand and one did, suggesting binding followed by a conformational change).
Conformational Selection
![]()
Induced Fit
![]()
P* in the conformational selection models represents a high affinity, pre-existing conformation of the protein. In this model, high ligand shifts the equilibrium to the right.
One way to differentiate these models is to look at the dependency of the different kinetic phases on ligand. In the conformation selection model, the slow step is the formation of the high affinity form of the protein, P*. The first slow step has a nonlinear dependence in L while the fast second step has a linear dependence. The data did not fit this model well.
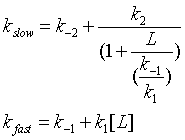
In the induced fit, the ligand binds to a low affinity and perhaps unfolded form of the protein, which subsequently collapses to the bound form in a slow step.
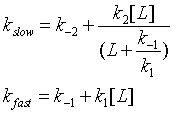
Both ligand dependent and independent phases are evident in the equation for the slow step for the induced fit mechanism. At high ligand concentration (when L >> k-1/k1) , the slow step in the induced fit would be independent of ligand (kslow = k-2 + k2). The authors state the data is consistent with a variant of induced fit called the "fly casting model". In this model the protein first encounters ligand and forms a hydrophobic collapse intermediate (PL) in a fast step characterized by a linear dependence on ligand concentration. Then the intermediate slowly interconverts into a wild type like complex through conformational re-arrangement. Wild-type protein binds the ligand 1000x as quickly, suggesting entropic barrier to binding of the ligand to the unfolded state and rearrangement of the protein thereafter.
Junker et al used atomic force microscopy (AFM) to observe the
effects of ligand binding on the folding/unfolding fluctuations of a single
molecule of calmodulin (CaM), a calcium-binding protein that binds
amphiphilic helicals peptides which leads to a large conformation change in
the protein. To do this, they sandwiched a single CaM molecule between
filamins that serve as attachment points for the AFM tip and a surface.
A slow pulling force was then applied to the molecule, and the length gain
was measured as the protein unfolded. The rapid fluctuations between
folded and unfolded states were quantified and used to derive a complete
energy landscape for the folding of CaM.
 Navigation
Navigation
Return to Chapter 5C: Model Binding Sections
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 5C: Model Binding Systems

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.