Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 7 - CATALYSIS
A: METHODS OF CATALYSIS
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 04/11/16
|
Learning Goals/Objectives for Chapter 7A: After class and this reading, students will be able to
|
A4. Intramolecular Catalysis (reference)
Consider the hydrolysis of phenylacetate. This reaction, a nucleophilic subsitution reaction, could be catalyzed by the addition to solution of the general base acetate, as described above. Since this reaction would double with the doubling of the solution acetate, the reaction is bimolecular (first order in reactant and catalyst). Now consider the same reaction only when the the general base part of the catalyst, the carboxyl group, is part of the reactant phenylacetate. Such a case occurs in the acetylated form of salicylic acid - i.e. aspirin. When the carboxy group is ortho compared to the acetylated phenolic OH, it is in perfect position to accept a proton from water, decreasing the charge development on the O in the transition state. The general base does not have to diffuse to the appropriate site when it is intramolecular with respect to the carbonyl C of the ester link. The rate of this intramolecular base catalysis is about 100 fold greater than of an intermolecular base catalyst like acetate. It is as if the effective concentration of the intramolecular carboxyl base catalyst is much higher due to its proximity to the reaction site.
Another type of reactions involving a carboxyl group (in addition to simple proton transfer) is when the negatively charged carboxyl O acts as a nucleophile and attacks an electrophilic carbonyl carbon. When the carbonyl is part of an ester, the carboxyl group engages in a nucleophilic substitution reaction, expelling the alcohol part of the ester as a leaving group. The remaining examples below consider the nucleophilic (carboxyl) substitution on phenylesters, with phenolate as the leaving group. The reactions in effect transfer an acyl group to the carboxyl group to create an anhydride.
First consider acyl transfer with aspirin derivatives. Aspirin, as you know, contains a carboxyl group ortho to an ester substitutent. Hence the carboxyl group can act as a nucleophile and attack the carbonyl carbon of the ester in a nucleophilic substitution reaction. The net effect is to transfer the acetyl group from the phenolic OH to the carboxyl group converting it to an anhydride. This is an intramolecular reaction. Compare this reaction to a a comparable bimolecular reaction shown below.
Figure: ACYL TRANSFER IN ASPIRIN DERIVATIVES.
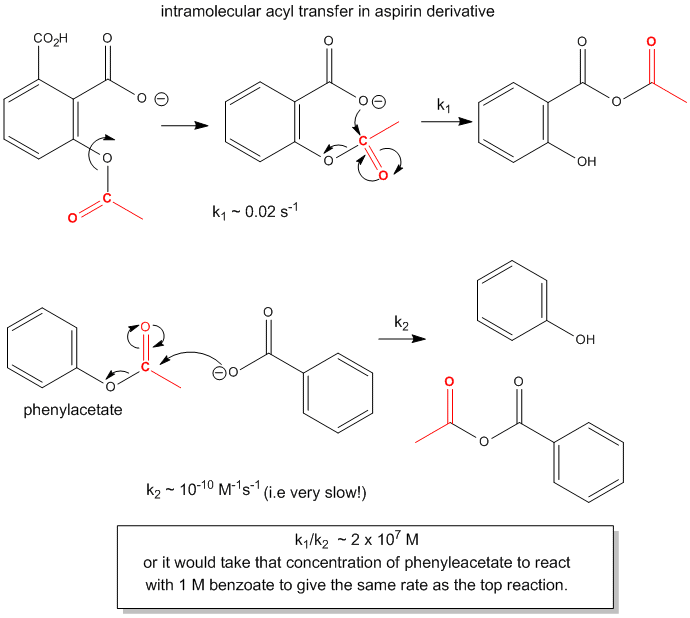
The first order rate constant of the intramolecular transfer of the acetyl group to the carboxyl group, k1 = 0.02 s-1. The analogous bimolecular reaction rate constant k2~ 10-10 M-1s-1. Dividing k1/k2 gives the relative rate enhancement of the intramolecular over the intermolecular reaction. With units of molarity, this ratio can be interpreted as the relative effective concentration of the intramolecular nucleophile. This makes the effective concentration of the carboxylate in the aspirin derivative 2 x 107 M.
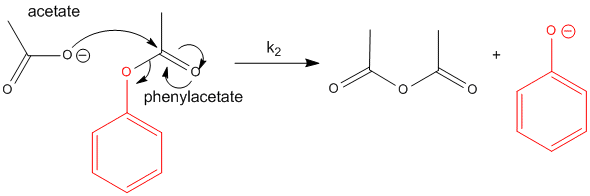
Now consider the cleavage of phenylacetate using acetate as the nucleophile. The products are acetic anhydride and phenolate. This is a bimolecular reaction (a slow one at that), with a bimolecular rate constant, k2 which I will arbitrarily set to 1 for comparison to some similar reactions.
Figure: REACTION OF ACETATE WITH PHENYLACETATE
Now consider a monoester derivatives of succinic acid - phenyl succinate - in which the free carboxyl group of the ester attacks the carbonyl carbon of the ester derivative.
Figure: INTRAMOLECULAR REACTION OF PHENYLSUCCIATE
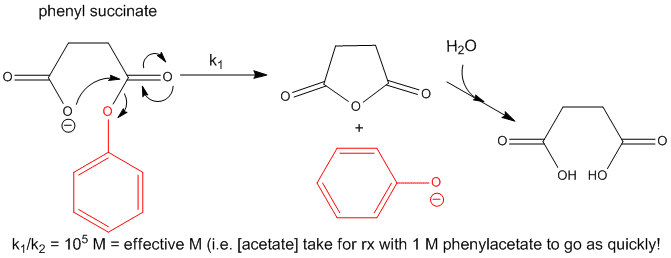
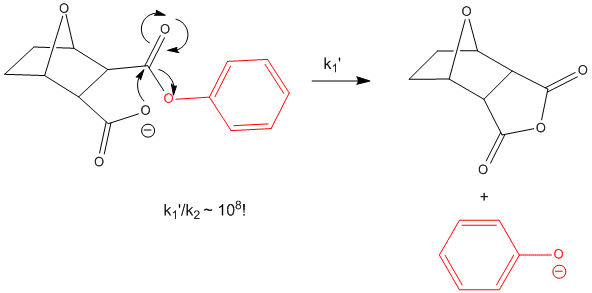
If you assign a second order rate constant k2 = 1 M-1s-1 to the analogous intermolecular reaction of acetate with phenylacetate (as described above), the first order rate constant for the intramolecular reaction of phenylsuccinate is 105 s-1. The ratio of rate constants, k1/k2 = 105 M. That is it would take 105 M concentration of acetate reacting with 1 M phenylacetate in the first bimolecular reaction to get a reaction as fast as the intramolecular reaction of phenylsuccinate. An even more sterically restricated bicyclic phenylcarboxylate shows a k1/k2 = 108 M.
Figure: INTRAMOLECULAR REACTION OF BICYCLIC PHENYLCARBOXYLATE
Another example is anhydride formation between two carboxyl groups. The ΔGo for such a reaction is positive, suggesting an unfavorable reaction. Consider two acetic acid molecules condensing to form acetic anhydride. For this intermolecular reaction, Keq = 3x10-12 M-1. Now consider the analogous intramolecular reaction of the dicarboxylic acid succinic acid. It condenses in an intramolecular reaction to form succinic anhydride with a Keq = 8x10-7 (no units). The ratio Keq-intra/Keq inter = 3 x 105 M. It is as if the effective concentration of the reacting groups. because they do not have to diffuse together to react, is 3 x 105 M.
How does this apply to enzyme catalyzed reaction? Enzymes bind substrates in physical steps which are typically fast. The slow step is chemical conversion of the bound substrate, which is effectively intramolecular. These three kinds of reactions, intermolecular, intramolecular, and enzyme-catalysed can be broken down into two hypothetical steps, a binding followed by catalysis.
Figure: three kinds of reactions, intermolecular, intramolecular, and enzyme-catalysed
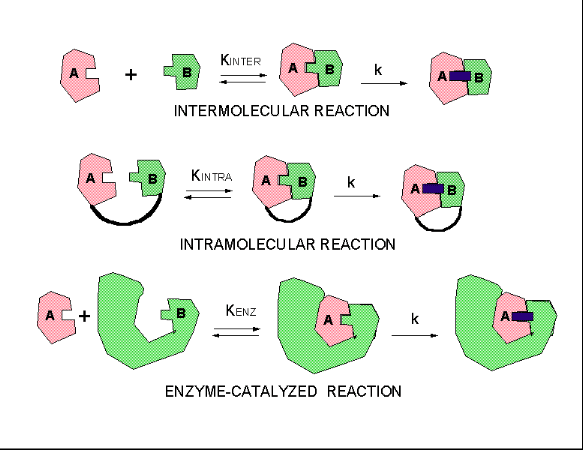
If the rate constants for the chemical steps are all identical, the advantage of the intramolecular and enzyme-catalyzed reaction over the intermolecular reaction is KINTRA/KINTER and KENZ/KINTER, respectively.
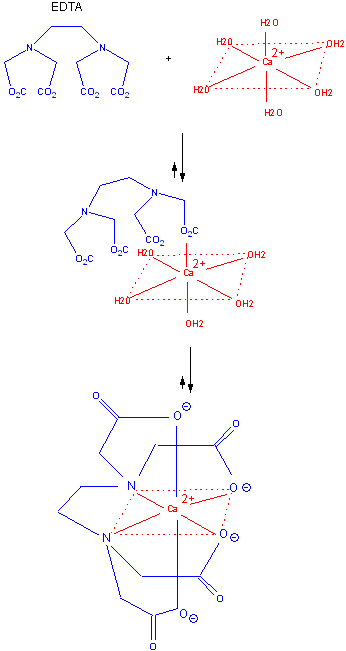
The advantage of intramolecular reactions can be seen by studying the Ca-EDTA complex. Calcium in solution exists as a octahedrally coordinated complex with water occupying all the coordination sites. EDTA, a multidentate ligand, first interacts through one of its potential six electron donors to Ca in a reaction which is entropically disfavored from the the Ca-EDTA perspective, although one water is released. Once this first intramolecular complex is formed, the rest of the ligands on the EDTA rapidly coordinate with the Ca and release bound water. The former is no longer entropically disfavored since it is now an intramolecular process while the later is favored through the release of the remaining five water molecules.
Figure: FIGURE: BINDING OF Ca2+ AND EDTA.
We modeled the catalytic advantage offered by intramolecular reaction in terms of a dramatic increase in the effective concentration of reactants, which sometimes reached levels of 108 M. Another way is to look at entropy changes associated with dimer formation. The table below shows that an intramolecular reaction is favored over an intermolecular reaction since in the latter, significant decreases in translational and rotation entropy result.
Translational, Rotational, and Internal Entropies
for Dimer Formation:
A + B <=> A-B (cal/K.mol)
| System | A | B | A-B | ΔS |
| Gas | ||||
| S trans | 30 | 30 | 30 | -30 |
| S rot | 20 | 20 | 20 | -20 |
| S int | 5 | 5 | 20 | +10 |
| Gas -> Solution | -10 | -10 | -15 | |
| S sol | 45 | 46 | 55 | -35 (Correspond to 108-109 M) |
 Navigation
Navigation
Return to 7A: Methods of Catalysis Sections
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 7A: Methods of Catalysis

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.