{kind=link}
Marshall, N. et al. Rationally tuning the reduction potential of a single cupredoxin beyond the natural range Nature 462, 113 (2009)
Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 8 - OXIDATION/PHOSPHORYLATION
B: OXIDATIVE ENZYMES
BIOCHEMISTRY - DR. JAKUBOWSKI
04/15/16
|
Learning Goals/Objectives for Chapter 8B: After class and this reading, students will be able to
|
B1. General Oxidizing Agents
Oxidizing agents are required to oxidize organic molecules. In organic lab, you never used dioxygen as an oxidizing agent. It is difficult to limit the extent of oxidation using dioxygen. In addition, side reactions are likely given the nature of the reactive oxygen reduction products. (The mechanisms of combustion reactions of organic molecules with dioxygen to produce carbon dioxide and water are very complicated.)
Figure: mechanisms of combustion reactions
|
INITIATION |
| CH4 --> CH3. + H . |
| O2 --> 2 O. |
|
PROPAGATION |
| CH4 + H . --> CH3. + H2 |
| CH4 + HO . --> CH3. + H2O |
| CH3. + O . --> CH2O + H. |
|
CH2O + HO . -> CHO. + H2O
|
| CH2O + H . -> CHO. + H2 |
| CHO. --> CO + H. |
| CO + HO . -> CO2 + H. |
|
BRANCHING |
|
H. + O2 --> HO. + O.
|
|
TERMINATION |
| H . + R. + M-> RH + M*
|
after Chemistry, 5th ed. Zumdahl. pg 1097
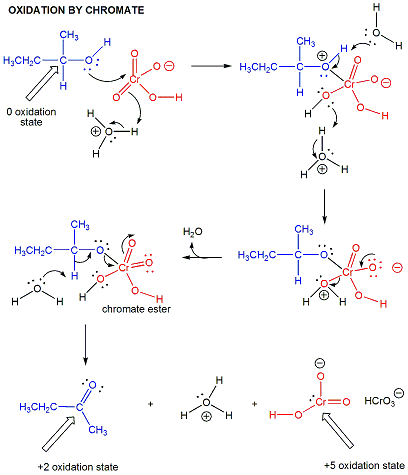
In organic lab, other oxidizing agents are often used, including permanganate and chromate.
Figure: permanganate
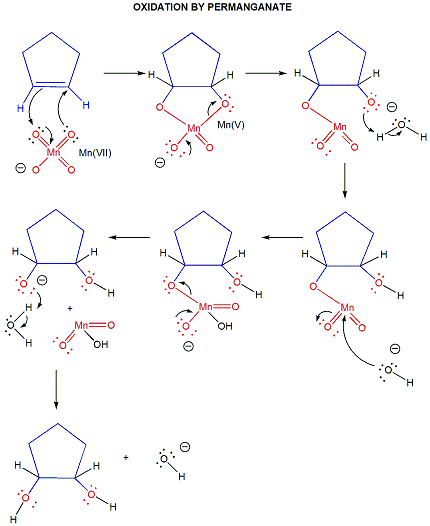
Figure: chromate
Oxygen can often be inserted into a molecule in a nonoxidative process by hydration of an alkene to an alcohol (a readily reversible reaction), which could then be oxidized to either an aldehyde/ketone or carboxylic acid using an appropriate oxidizing agent.
Most biological oxidation reactions (such as those found in glycolysis, Kreb Cycle, and fatty acid oxidation) do not use dioxygen as the immediate oxidizing agent. Rather they use nicotinamide adeninine dinucleotide (NAD+) or flavin adenine dinucleotide (FAD) as oxidizing agents, which get reduced. Enzymes that uses these oxidizing agents are usally called dehydrogenases. Dioxygen can also be used to introduce oxygen atoms into biological molecules in oxidative reactions. Enzymes that introduce one oxygen atom of dioxygen into a molecule (and the other oxygen into water) are called monooxygenases. (Note: some monooxygenase that hydroxylate biomolecules are called hydroxylases.) Those that introduce both atoms of dioxygen into a substrate are called dioxygenases. These oxygenases are not usually used to oxidize organic molecules for energy production. Rather they introduce O atoms for other reasons, including increasing the solubility of nonpolar aromatics to facilitate secretion, and to produce new molecular species which have different biological activities. Finally, biological molecules can be oxidized by dioxygen in which no atoms of oxygen are added to the substrate. Rather, electrons lost from the oxidized substrate are passed via intermediate electron carriers to dioxygen , which get reduced to superoxide (if one electron is added), hydrogen peroxide (if two electrons are added) or water (if 4 electrons are added). These enzymes are called oxidases. (Note: The letters oxi- or oxygen- are used in all the enzymes that use dioxygen as the oxidizing agent.)
In this chapter section, we will discuss biological oxidation reactions. Most introductory biochemistry texts don't approach oxidation reactions in one cohesive chapter. Probably because of that, when I was learning biochemistry, I found the presentation of these different enzymes involved in redox reactions to be very confusing. Hopefully this section will alleviate that problem. First the chemistry of NAD+ and FAD will be discussed. Then the enzymes using dioxygen in oxidative reactions (monooxygenases, dioxygenases, and oxidases) will be explored.
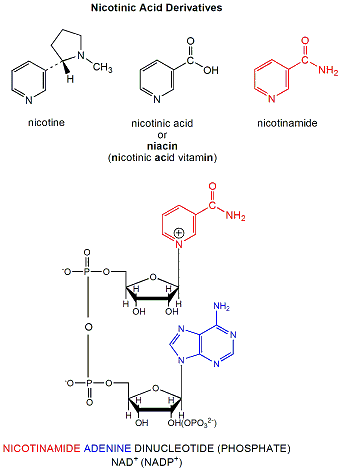
B2. The Chemistry of NAD+ and FAD
NAD+ is a derivative of nicotinic acid or nicotinamide.
Figure: NAD+ is a derivative of nicotinic acid or nicotinamide.
It and its reduction product, NADH, exists in the cells as interconvertible members of a pool whose total concentration does not vary significantly with time. Hence, if carbohydrates and lipds are being oxidized by NAD+ to produce energy in the form of ATP, levels of NAD+ would begin to fall as NADH rises. A mechanism must be be present to regenerate NAD+ from NADH if oxidation is to continue. As we will see later, this happens in the muscle under anaerobic conditions (if dioxygen is lacking as when you are running a 100 or 200 m race, or if you are being chased by a saber-toothed tiger) when pyruvate + NADH react to form lactate + NAD+.
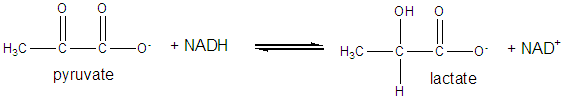
Under aerobic conditions (sufficient dioxygen available), NADH is reoxidized in the mitochondria by electron transport through a variety of mobile electron carriers, which pass electrons to dioxygen (using the enzyme complex cytochrome C oxidase) to form water.
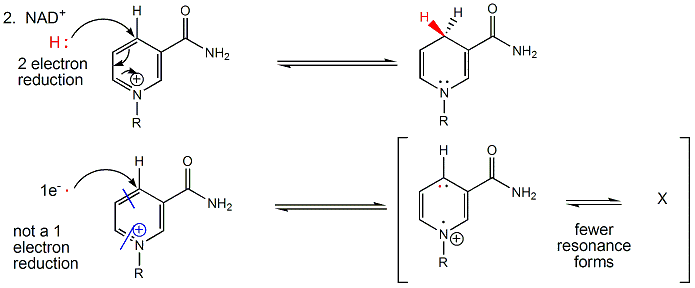
NAD+/NADH can undergo two electron redox steps, in which a hydride is transferred from an organic molecule to the NAD+, with the electrons flowing to the positively charged nitrogen of NAD+ which serves as an electron sink. NADH does not react well with dioxgyen, since single electron transfers to/from NAD+/NADH produce free radical species which can not be stabilized effectively. All NAD+/NADH reactions in the body involve 2 electron hydride transfers.
Figure: All NAD+/NADH reactions in the body involve 2 electron hydride transfers
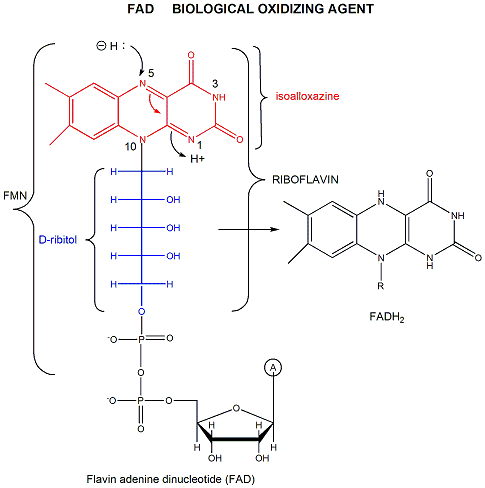
FAD (or flavin mononucleotide-FMN) and its reduction product, FADH2, are derivatives of riboflavin.
Figure: derivatives of riboflavin
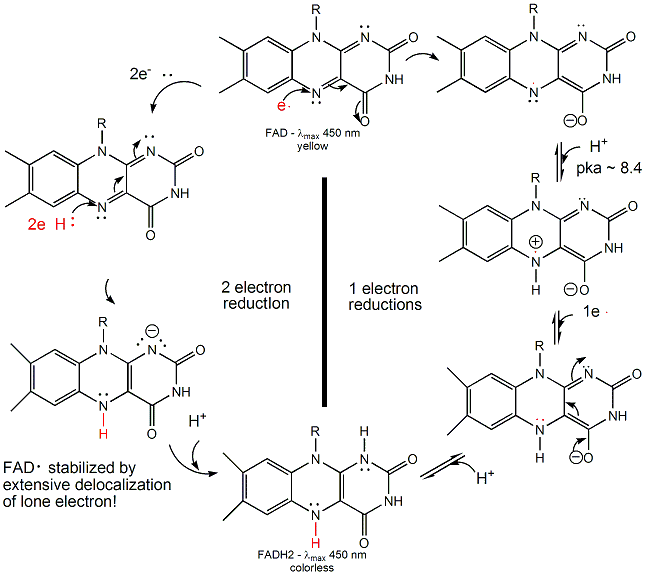
FAD/FADH2 differ from NAD+/NADH since they are bound tightly (Kd approx 10-7 - 10-11 M) to enyzmes which use them. This is because FADH2 is susceptible to reaction with dioxygen, since FAD/FADH2 can form stable free radicals arising from single electron transfers. FAD/FADH2 can undergo 1 OR 2 electrons transfers.
Figure: FAD/FADH2 can undergo 1 OR 2 electrons transfers
FAD/FADH2 are tightly bound to enzymes so as to control the nature of the oxidizing/reducing agent that interact with them. (i.e. so dioxygen in the cell won't react with them in the cytoplasm.) If bound FAD is used to oxidize a substrate, the enzyme would be inactive in any further catalytic steps unless the bound FADH2 is reoxidized by another oxidizing agent.
B3. DEHYDROGENASES
These enzymes usually involve NAD+/NADH and are named for the substrate
that is oxidized by NAD+. For instance in the reaction:
pyruvate + NADH <===> lactate + NAD+
which is used to regenerate NAD+ under anerobic conditions, the enzyme is named lactate dehydrogenase. As in acid/base reactions, when the preferred direction for the reaction (from a ΔGo perspective) is from stronger acid to weaker (conjugate) acid, the preferred direction for a redox reaction is in the direction from strong to weak oxidizing/reducing agents. This can easily be determined from charts of standard reduction potentials, and using the equation: ΔGo = -nFΔEo,
- where F is the Faraday constant (96,494 Coulombs/mol e- = 96, 494 J/(V.mol) = 23.06 kcal/(V.mol) . One Faraday is the charge per one mol of electrons).
- andΔEo, the standard EMF or standard cell potential (total voltage at standard state conditions), which can be determined by adding the standard reduction potentials (Eo) for the two appropriate half-reactions, after reversing the equation for the half-reaction that represents the oxidation.
When n=2 (number of electrons) which is common for oxidations of organic molecules,
ΔGo (kcal/mol) = - 46.12ΔEo or for government work ΔGo (kcal/mol) = - 50ΔEo
Notice when ΔEo > 0, ΔGo < 0, the reaction as written is favored under standard conditions. Note in the table below that many of the half reactions involve protons. For biological reactions involving free protons, the standard state concentration for the protons are not 1 M as for other solutes in solution, but defined to be the hydronium ion concentration at pH 7.0. The ΔEo and ΔGo values for the reactions involving hydrogen ions at a standard state of pH 7.0 are usually written as ΔEo' and ΔGo'
Table: Standard Reduction Potential Table (E0'), 25oC
| oxidant |
reductant |
n (electrons) | Eo� (volts) |
| Acetate + carbon dioxide |
pyruvate |
2 | -0.70 |
| succinate + CO2 + 2H+ |
α−ketoglutarate + H2O |
2 | -0.67 |
| acetate |
acetaldehyde |
2 | -0.60 |
| glycerate-3-P | glyceraldehyde-3-P + H2O | 2 | -0.55 |
| O2 | O2- | 1 | -0.45 |
| ferredoxin (ox) | ferredoxin (red) | 1 | -0.43 |
| Carbon dioxide |
formate |
2 | -0.42 |
| 2H+ |
H2 |
2 | -0.42 |
| α-ketoglutarate + CO2 + 2H+ |
isocitrate |
2 | -0.38 |
| acetoacetate | β-hydroxybutyrate | 2 | -0.35 |
| Cystine |
cysteine |
2 | -0.34 |
| Pyruvate + CO2 |
malate |
2 | -0.33 |
| NAD+ + 2H+ |
NADH + H+ |
2 | -0.32 |
| NADP+ + 2H+ |
NADPH + H+ |
2 | -0.32 |
| FMN (enzyme bound) | FMNH2 | 2 | -0.30 |
| Lipoic acid, ox |
Lipoic acid, red |
2 | -0.29 |
| 1,3 bisphosphoglycerate + 2H+ |
glyceraldehyde-3-P + Pi |
2 | -0.29 |
| Glutathione, ox |
red |
2 | -0.23 |
| FAD (free) + 2H+ |
FADH2 |
2 | -0.22 |
| Acetaldehyde + 2H+ |
ethanol |
2 | -0.20 |
| Pyruvate + 2H+ |
lactate |
2 | -0.19 |
| Oxalacetate + 2H+ |
malate |
2 | -0.17 |
| α-ketoglutarate + NH4+ |
glutamate |
2 | -0.14 |
| FAD + 2H+ (bound) |
FADH2 (bound) |
2 | 0.003-0.09 |
| Methylene blue, ox |
Methylene blue, red |
2 | 0.01 |
| Fumarate + 2H+ |
succinate |
2 | 0.03 |
| CoQ (Ubiquinone - UQ + H+ | UQH. | 1 | 0.031 |
| UQ + 2H+ | UQH2 | 2 | 0.06 |
| Dehydroascorbic acid |
ascorbic acid |
2 | 0.06 |
| Ubiquinone; ox |
red |
2 | 0.10 |
| Cytochrome b2; Fe3+ |
Cytochrome b2; Fe2+ |
1 | 0.12 |
| Cytochrome c1; Fe3+ |
Cytochrome c1; Fe2+ |
1 | 0.22 |
| Cytochrome c; Fe3+ |
Cytochrome c; Fe2+ |
1 | 0.25 |
| Cytochrome a; Fe3+ |
Cytochrome a; Fe2+ |
1 | 0.29 |
| 1/2 O2 + H2O |
H2O2 |
2 | 0.30 |
| Cytochrome a3; Fe3+ |
Cytochrome a3; Fe2+ |
1 | 0.35 |
| Ferricyanide |
ferrocyanide |
2 | 0.36 |
| Cytochrome f; Fe3+ |
Cytochrome f; Fe2+ |
1 | 0.37 |
| Nitrate |
nitrite |
1 | 0.42 |
| Photosystem P700 | . | . | 0.43 |
| Fe3+ |
Fe2+ |
1 | 0.77 |
| 1/2 O2 + 2H+ |
H2O |
2 | 0.816 |
-
 Recommendations
for nomenclature and tables in biochemical thermodynamics (from the
International Union of Biochemistry and Molecular Biology and the IUPAC)
Recommendations
for nomenclature and tables in biochemical thermodynamics (from the
International Union of Biochemistry and Molecular Biology and the IUPAC)
The mechanism for the oxidation of a substrate by NAD + involves concerted hydride transfer to one face of NAD+.
Consider for example the oxidation of ethanol to acetaldehyde by alcohol dehydrogenase.
Figure: oxidation of ethanol to acetaldehyde by alcohol dehydrogenase
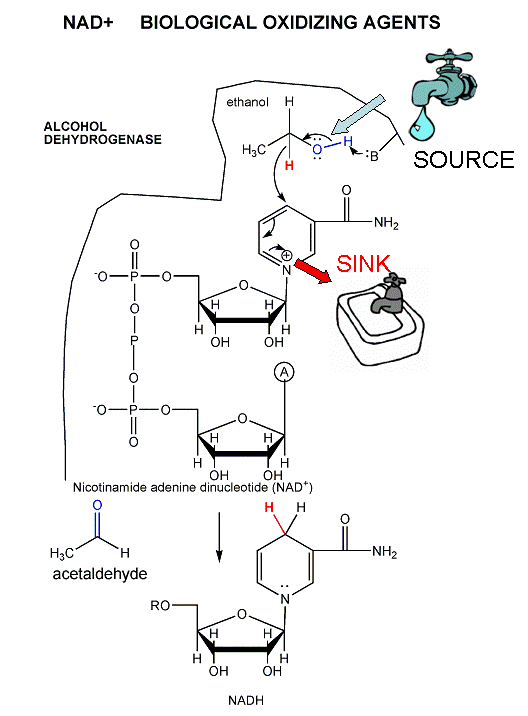
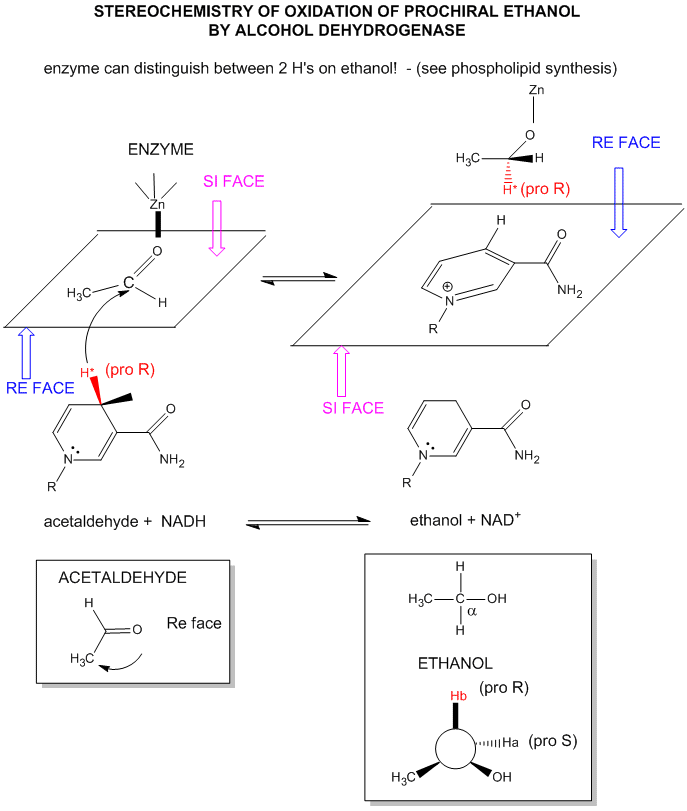
For substrates like ethanol that loses a hydride from a methylene carbon atom that has two H's, only one of the H's is lost (either the proR or proS) from the prochiral center. (Remember the reaction of prochiral glycerol to give phospholipids.)
Figure: STEREOCHEMISTRY OF NAD+/NADH REDOX REACTIONS WITH ALCOHOL DEHYDROGENASE
FAD has a more positive reduction potential than NAD+ so it is used for more "demanding" oxidation reactions, such as dehydrogenation of a C-C bond to form an alkene. You will notice on standard reduction potential tables that the potential of FAD is often listed several times and depends on the enzyme. This is because the FAD is tightly bound to the enzyme so its tendency to acquire electrons depends on its environment, in much the same fashion as the pKa of an amino acid side chain (which reflects is tendency to release protons) is affected by the environment of the amino acid side chain in the protein. The standard reduction potential for flavin enzymes varies from -465 mV to + 149 mV. Compare this to the reduction potential of free FAD/FADH2, which in aqueous solution is -208 mV. The standard reduction potential of the flavin in D-amino acid oxidase, a flavoprotein, is about 0.0 V. Remember, the more positive the standard reduction potential, the more likely the reactant will be reduced and hence act as an oxidizing agent. Hence the FAD in D-amino acid oxidase is a better oxidizing agent than free FAD. The Kd for binding of FAD to the enzyme is 10-7M compared to the Kd for binding of FADH2, which is 10-14M. By gaining electrons, the flavin binds more tightly, which preferentially stabilizes the bound FADH2 compared to the bound FAD. This shifts the equilibrium of FAD <=> FADH2 to the right, making the bound FAD a stronger oxidizing agent.
Figure: FAD AND OXIDATIONS: MECHANISM
![]() Jmol:
Updated Flavin dehydrogenase
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Flavin dehydrogenase
Jmol14 (Java) |
JSMol (HTML5)
Can the standard reduction potential of a redox active center in a protein be tuned by changing the environment of that center, much as the pKa of an acid side chain can by changing the polarity of the environment? The answer is yes. The active site of azurin, a cupredoxin, has a redox active copper ion coordinated by a Cys and two His residues in a trigonal planar fashion. Met 121 serves as a weak axial ligand. Marshall et al. have reported a feasible method to manipulate the redox potential (Eo) of this active site. The wild type azurin was mutated to alter the hydrophobicity and hydrogen bonding capabilities, while maintaining the overall architecture of the metal binding site. Ser 46 was selected for mutation since it occupied a position similar to Asn in another curedoxin that was involved in an important H bond binding two ligand binding loops. An N47S-mutation, which strengthened the hydrogen bond between the two ligand-containing loops increased Eo by ~130 mV while preserving metal binding site architecture as determined by UV-Vis spectroscopy. They also compared a M121Q mutant with wild-type M121 and with a M121L mutant. A plot of Eo vs log partition coefficient for transfer of the side chain from water to octanol was essentially linear with a positive slope, showing that the standard reduction potential depended on the hydrophobicity of the weakly coordinating ligand in the metal binding region. This behavior extended to double mutants (where one set of mutants involved M121). The investigators were able to tune the Eo over a 700 mv range! New Role for NAD.
B4. Another role of NAD - Sirtuins
Investigations of aging in many organisms have shown that a plethora of proteins seem to be involved. Studies have shown that overexpression of the yeast Sir2 (silence information regulator) caused an increase in lifespan. The enzyme product of this gene appear to be involved in inhibition, through covalent modification of chromatin and associated gene silencing, of genetic recombination. Investigators have shown that this protein is a NAD+-dependent histone deacetylase. Sir2 is a member of a large family of conserved protein deacetylases called sirtuins. Accompanying the deacetylase activity is the conversion of NAD+ to nicotinamide and acetyl-ADP ribose. Since the enzyme uses NAD+, its activity would be related to the metabolic state of the cell. It was thought that high levels of NAD+ would be present when metabolism was slow, and these high levels of NAD+, through its activation of Sir2, would lead to the silencing of genes and longer lifespan. Lack of genetic recombination would decrease DNA damage. Alternatively, low levels of nicotinamide (a product of Sir2) could minimize inhibition of Sir2 and raises its activity. However, Anderson et al. reported that in yeast, caloric restriction actually reduces NAD+, and that the activity of Sir2 does not depend on the ratio of NAD+/nicotinamide, suggesting that Sir2 is activated (during caloric restriction) by other molecules.
It has long been know that caloric restriction increases lifespans in a variety of animals. This provides a mechanism that would account for that. When glucose levels in culture media are restricted, the life span of the yeast increases, as measured by an increase in the reproductive life span of the organism. This effect is only observed if a functional Sir2 gene is present. Deacetylation of histone proteins by Sir2, would lead to an increased positive charge on the histones (protein components of the nucleosome), which presumably increases the binding affinity of histones (in nucleosomes) for DNA. This could reduce gene expression (i.e. induce silencing) since the nucleosomal DNA would be less available for transcription. Surprisingly, a precursor of cellular NAD+, nicotinamide, when given to cells to increase NAD+ levels which should silence gene expression had the opposite effect. The effect of Sir2 on life span has also been observed in a more complex organism, C. Elegans (round worm).
Another activity of Sir2 in yeast has been noted by Aguilaniu et al. When yeast cells divide, daughter cells do not inherit small DNA circles which code for ribosomal RNA. The concentration of these circles of DNA increase with age, and are associated with senescence of the maternal cells. Cells without Sir2 have more of these circles. Another feature of aging cells is the accumulation of oxidized proteins (as evidenced by an increase in carbonyl groups). In a fashion similar to rDNA circles, oxidatively-modified proteins are not inherited by daughter cells except in mutant yeast cells lacking Sir2.
Since increasing levels of Sir2 increase lifespan, ligands that bind to existing Sir2 and activate its deacetylase activity might also increase lifespan. Howitz et al. screened libraries of compounds for such ligands and found one exceptionally good one that activated the enzyme 13-fold: resveratrol, a polyphenol found in red wine.
Figure: resveratrol
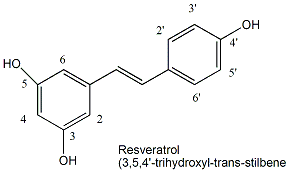
Studies have shown the people who drink moderate amounts of red wine may have lower risk for certain disease (for example some cancers and cardiovascular disease) and longer life spans. When given to yeast, resveratrol increased their life span by 70% in a process that required Sir2. Mutant strains lacking Sir 2 did not accrue the effect. Whether resveratrol levels found in wine are sufficient to promote this effect in humans is unclear. Wood et al. have recently extended the effects of resveratrol on activation of Sir2 and increased life-span to more complex organism - C. elegans and Drosophila melanogaster.
In humans there appears to be seven different sirtuins. Sirtuin 2 has been shown to deacetylate α-tubulin (involved in cycle cycle), p53 (a tumor suppressor gene) and histones H3 and H4. In some cases inhibition of sirtuin activity is beneficial.
The product of the gene DBC1 (deleted in breast cancer 1) has been shown to bind to and inhibit SIRT1, which causes increases in the concentration of acetylated p53 and other proteins regulated by it. One function of active p53 is to induce cell death (apotosis) in stressed cells (such as cancer cells). Hence sirutin 1 would inhibit p53 induced apotosis of cancer cells. .
Figure: Reaction of NAD+ and Acetyl-Lys side chains: possible catalytic mechanism for yeast Sir2
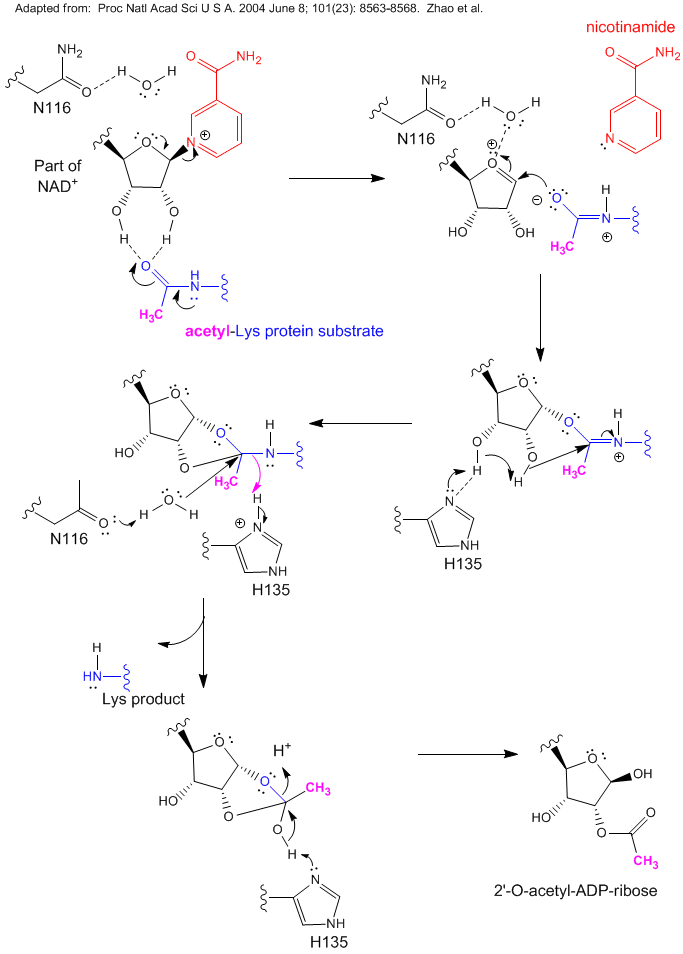
The effects of resveratrol has been extended to mammals through the study of its effects on mouse Sir1, one of seven similar proteins in mammals, is analogous to Sir2 in yeast. Sir1 is also a deacetylase which appears to work on many different acetylated proteins. It functions in part with another protein, peroxisome proliferator-activated receptor γ coactivator (PGC-1α), to regulate two important carbohydrate metabolism pathways in the liver under conditions of caloric restriction, probably through responses to NAD+ levels. The two pathways are glycolysis, the breakdown of the 6C glucose to the 3C molecule pyruvate, and gluconeogenesis, the synthesis of glucose from pyruvate. When blood sugar (glucose) is low, or in a diabetic state (when blood glucose levels are high due to impaired insulin function which facilitates glucose uptake into cells), sugar metabolism in the liver shifts to gluconeogenesis to produce glucose for export. This might sound contradictory in the diabetic state when blood glucose is abundant. However, the liver is responding to other signals in the blood sent from tissues which lack glucose, so its response is to synthesis glucose and release it into the blood. Sir1 deacetylates acetylated Lys side chains on PGC-1α, which increases its biological function and leads to transcription of liver genes for gluconeogenesis.
Two studies in mice extent the beneficial effects of resveratrol. In one study by Baur et al., mice fed a high calorie diet and who became obese avoided the negative effects of obesity if given moderate amounts of resveratrol. Compared to controls who were fed the same diet and not given resveratrol, the treated mice had a much longer life expectancy. Resveratrol appears to alter the deleterious changes in gene expressions in mice that are associated with insulin resistance, diabetes and cardiovascular disease. Resveratrol increased insulin sensitivity, decreased a hormone called insulin-like growth factor 1 (IGF-1, increased PGC-1αactivity, and mitochondrial numbers.
In another study by Lagouge et al, ordinary (untrained) mice given resveratrol had markedly increased endurance and oxygen consumption with decreased heart rate on a treadmill test, suggesting improved mitochondrial function. Resveratrol increased transcription of muscles (not liver) genes associated with mitochondrial oxidative phosphorylation (the topic of the next section) and with production of new mitochondria (mitochondria biogenesis). They studied mitochondrial function since this organelle is the site of most oxygen consumption and presumably the most reactive oxygen species (ROS), which have a role in aging and disease. Mitochondrial function also decreases with age. The effects of resveratrol on one particularly important gene involved in mitochondrial function, PGC-1α, was noted. Transcription of this gene in muscle is decreased in diabetics and in people resistant to insulin, both of which suffer impaired mitochondrial function. This protein has many biological functions, including increasing mitochondrial function and biogenesis. As noted above, resveratrol decreased acetylation of PGC-1α, and increased its function, which in muscle lead to increased transcription of genes involved in mitochondrial biogenesis an oxidative phosphorylation. They also showed that these effects were mediated through the Sir1 protein since PGC-1 acetylation was not affected by resveratrol in mutant mouse embryonic fibroblast cells with a nonfunctional SIR1 gene.
B5. Hydrogenases (not dehydrogenases): A break from oxidation reactions
Our world desperately needs an energy efficient way to produce H2 for energy production without producing waste pollutants. Catalytic cracking of molecules and newly developed fuel cells offer two possibilities. Wouldn't it be great if a reactant like water could be used for H2 production (without the use of electrolysis) or expensive metal catalysts? Nature may show the way. Bacteria (even E. Coli found in our GI system) can use simple metals like iron to produce H2 from H+ with electrons for the reduction of H+ coming from a donor (such as a reduced heme in proteins):
Dred+ H+ <=> Dox + H2
The reaction is also reversible in the presence of an acceptor of electrons from H2 as it gets oxidized:
Aox+ H2 <=> Ared + H+
The enzymes that catalyze hydrogen production are hydrogenases (not dehydrogenases). Note that the name hydrogenases best reflects the reverse reaction when a molecule (P) in an oxidized state gets reduced (to S) and H2 gets oxided to H+.
Crystal structures of hydrogenases show them to be unique among metal-containing enzymes. They contain two metals bonded to each other. The metal centers can either be both iron or one each of iron and nickel. The ligands interacting with the metals are two classical metabolic poisons, carbon monoxide and cyanide. Passages for flow of electrons and H2 connect the buried metals and the remaining enzymes. The metals are also bound to sulfhydryl groups of cysteine side chains. It appears that two electrons are added to a single proton making a hydride anion which accepts a proton to form H2. In the two Fe hydrogenases, the geometry of the coordinating ligands distorts the bond between the two iron centers, leading to irons with different oxidation numbers. Electrons appear to flow from one center to the other, as does carbon monoxide as well. Ultimately, hydrogenases or small inorganic mimetics of the active site could be coated on electrodes and used to general H2 when placed in water in electrolytic experiments.
B6. Monooxygenases
An examples of monooxygenases are the hydroxylases which hydroxylate amino acids like Trp and Tyr to form 5-hydroxytrytophan and 3-4-dihydroxyphenylalanine or dopa, respectively. These latter two substances can be decarboxylated using PLP-dependent enzymes to form the neurotransmitters 5 hydroxytryptamine (5HT or serotonin) and dopamine. The latter can be hydroxylated again to form norepinhephrine, and subsequently methylated to form epinephrine. LSD and amphetamine are anaologs of serotonin and dopamine, respectively.
Figure: DIAGRAM: TRP AND TYR CHEMISTRY
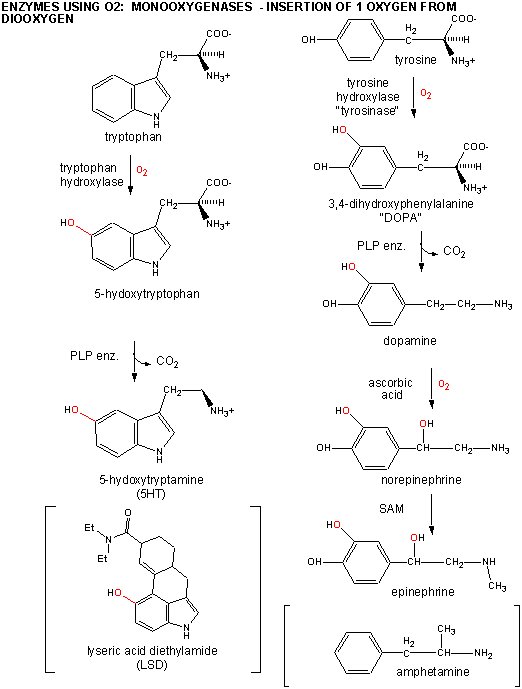
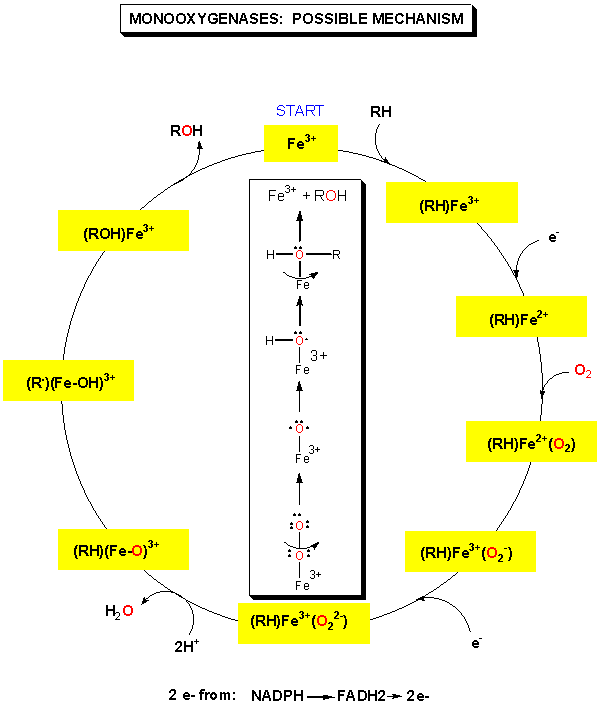
Since these monooxygenases use dioxygen, you might expect that the enzymes would use the motifs described in the previous section to facilitate its reaction with dioxygen. In fact, the enzyme contains a metal ion (Fe2+) bound to a heme in the protein. In addition, the reduction products of dioxygen that are eventually used to hydroxylate the substrate stay bound to the enzyme.
Figure: MONOOXYGENASES: POSSIBLE MECHANISM
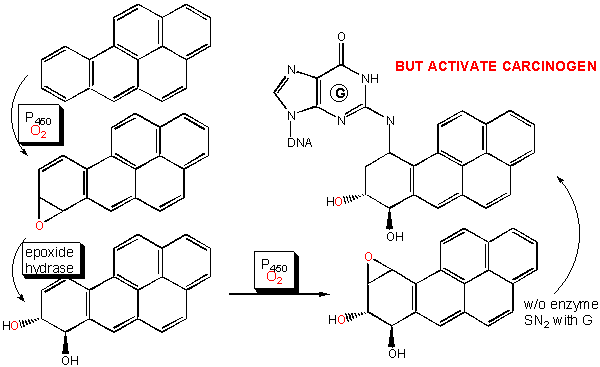
An important class of monoxygenases are called cytochromes P450's. They represent a class of similar enzymes that each contain a heme. Instead of reversibly carrying dioxygen as does the heme in myoglobin and hemoglobin, the P450 heme activates dioxgyen for hydroxylation reactions involving aromatic, nonpolar substrates. Hydroxylation of these substrates increases their solubility which facilitates their elimination from the body. Epoxide intermediates or products are often produced, which can open up through nucleophilic attack using an alcohol (sugar derivative) or an amine (such as in nucleotide bases in DNA) to form large adducts. Hence cytochrome P450 can actually activate aromatic substrates to become carcinogens.
Figure: CYTOCHROME P450'S: POSSIBLE MECHANISM
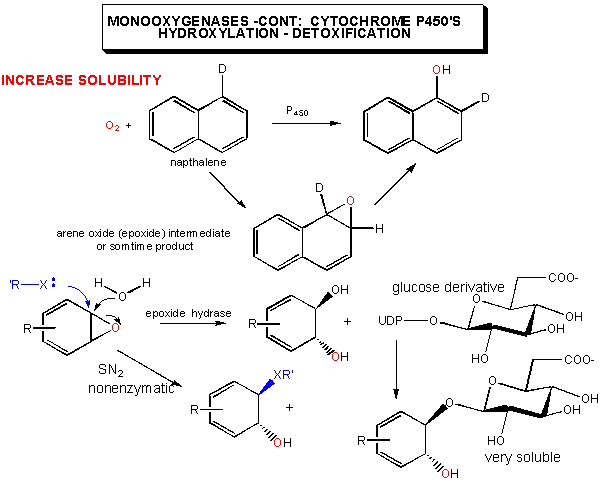
Figure: CYTOCHROME P450'S: ACTIVATION OF CARCINOGENS
The cytochrome P450s family of genes/proteins are inducible on exposure to nonpolar aromatic molecules such as dioxin. These nonpolar molecules can enter the cytoplasm where they bind to the arylhydrocarbon receptor (AhR) which is bound to a heat shock protein, Hsp90. Upon binding of dioxin, TCDD, for example, the AhR.TCCD complex dissociates from Hsp90, and migrates to the nucleus where it binds a protein called Amt. The AhR-Amt complex serves as an enhancer/transcription factor, facilitating the transcription of the cytochrome P450 genes.
Figure: Cytochrome P450s inducible on exposure to nonpolar aromatic molecules such as dioxin.

Dioxin has been shown to affect estrogen-mediated activities. Estrogens, small hydrophobic hormones derived from cholesterol, enter cell and bind to cytoplasmic estrogen receptors, which then dimerize and bind to the estrogen response element (ERE), initiating transcription. Recently the crystal structure of the ligand binding domain of the estrogen receptor, bound to tamoxifen, was reported. Tamoxifen, a drug derived from the yew plant, blocks the biological effects of the estrogen receptor. Although it binds to the estrogen receptor, it doesn't elicit the same conformational changes in the protein, which prevents the bound receptor from binding to the estrogen response element and recruiting other proteins needed for estrogen-dependent gene transcription. It is used in chemotherapy and prevention of estrogen-dependent breast cancer cells.
![]() Jmol: Updated
Estrogen Receptor:Tamoxifen Complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
Estrogen Receptor:Tamoxifen Complex
Jmol14 (Java) |
JSMol (HTML5)
How does dioxin interfer with estrogen signaling? As mentioned above, dioxin binds to the arylhydrocarbon receptor (AhR). This complex can then bind to Arnt - arylhydrocarbon nuclear transporter (a chaperone). (Note: unclear about AMT, Arnt relationship. Will fix soon). This ternary complex can then bind to the xenobiotic response element (XRE). Ahr and ARnt contain a basic helix-loop-helix motif which mediate their interaction with DNA. Upon binding of the complex, detoxification genes are activated. Ohtake et al. found that the dioxin-Ahr-Arnt complex can bind to the estrogen receptor, which can then lead to activation of the ERE in the absence of estrogen. However, if estrogen is present, inhibition of gene expression from ERE is observed. Dioxins can be potent disregulators of estrogen-induced gene expression. Such changes in esterogen activity could help to explain the pro- and inhibitory effects of dioxin on estrogen-mediated cellular responses and possible effects of dioxin on the immune system and on cancer development.
![]() EPA
Dioxin Reassessment: Risk
Characterization - Dose Response
EPA
Dioxin Reassessment: Risk
Characterization - Dose Response
![]() EPA
Dioxin Reassessment: Risk
Characterization - Mechanisms
EPA
Dioxin Reassessment: Risk
Characterization - Mechanisms
Recently, "snapshots" of P450cam, the cytochrome P450 that hydroxylates camphor, have been taken in various stages of catalysis. This enzyme is "the biological equivalent of a blowtorch: P450 enzymes catalyze the stereospecific hydroxylation of nonactivated hydrocarbons at physiological temperature - a reaction that, uncatalyzed, requires extremely high temperatures to proceed, even nonspecifically". Crystal structures of normal intermediates (such as the dioxygen-bound intermediate, could not be determined with traditional techniques since the complex had a half-life of 10 minutes at 4oC. This problem was solved by using crystals frozen at -185oC, and by using short wavelength X-rays, which did not cause the reaction to be driven forward. Short-time X-ray data was collected, then substrate added to push the reaction to the next intermediate, before new structural data was obtained.
Figure: Reaction Pathyway of P450cam
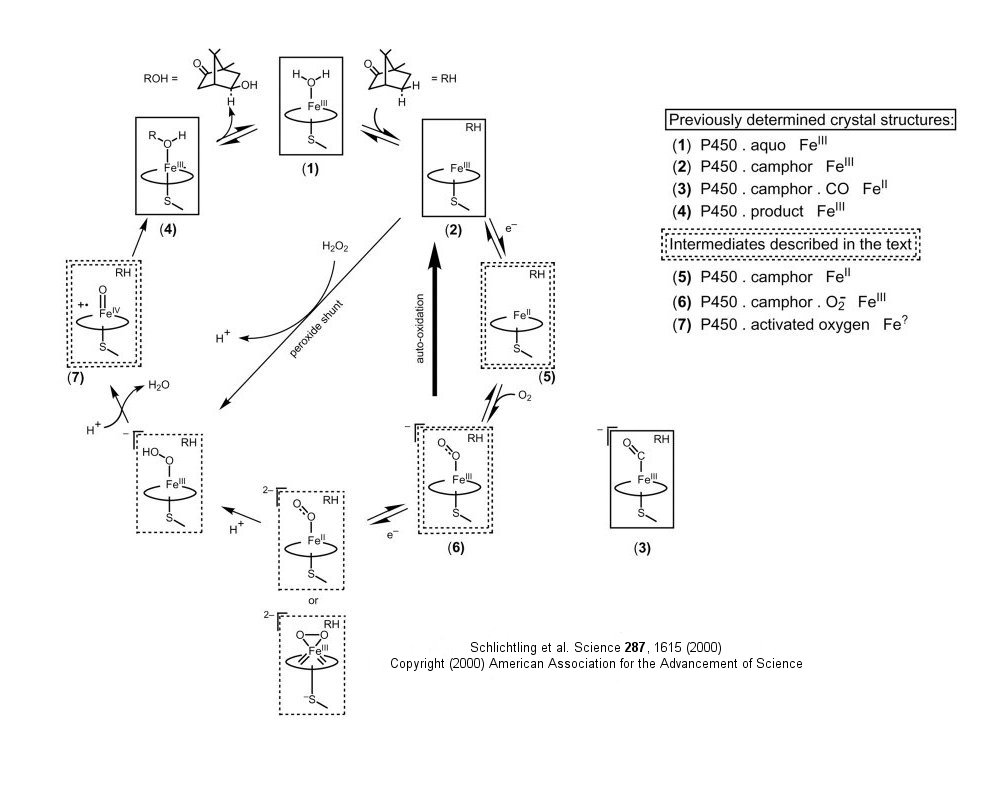
![]() Jmol:
Updated Cytochrome P450cam
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Cytochrome P450cam
Jmol14 (Java) |
JSMol (HTML5)
B7. Dioxygenases
An example of a dioxygenase is the cyclooxygenase activity of prostaglandin synthase. This enzyme, often just called cycloxygenase or COX, is an integral membrane protein found in the ER membrane, and is a homodimer (with two hemes). It catalyzes two different reactions. One is the addition of two dioxygens to arachidonic acid - 20:4Δ5, 8, 11, 15(which is liberated from the C2 position of phospholipid membranes by phospholipase A2 upon appropriate signaling) to form prostaglandin PGG2. This molecule, with 5 chiral centers, arises from arachidonic acid, which has one. The cyclooxygenase activity is buried in the membrane, from where the arachidonic acid can readily access its active site. The active site is at the end of a hydrophobic channel (arachidonic acid binding site) and stretches from the membrane-binding region to a buried heme. PGG2 can be further metabolized to PGH2 by the addition of two electrons to PGG2 by the hydroperoxidase activity of the enzyme, located at the other end of the enzyme. This activity forms an alcohol from the peroxide functional group in PGG2. There is one heme per momomer, which acts in both the cyclooxygenase and peroxidase activities. Each monomer of the dimer has both enzymatic activities.
Figure: Arachadonic Acid Bound to the Active Site of Mouse Cyclooxygenase
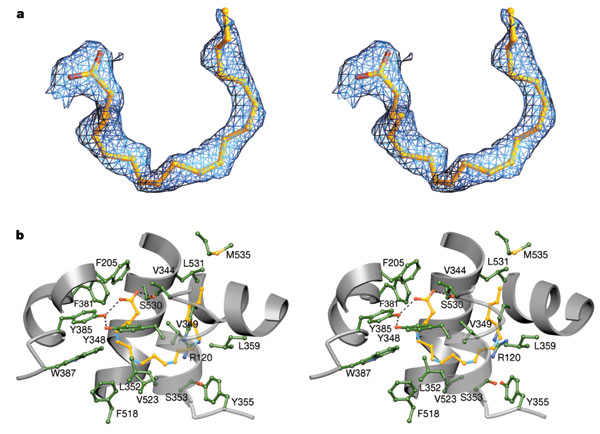
Reprinted by permission from Nature
Kiefer et al. Nature 405, 97-101
(2000)
Nature
Arachidonate and prostaglandin bound to the cyclooxygenase active site of wild-type COX-2. a, Stereo image of the experimental difference electron density map before the inclusion of AA (green trace) or PGH2 (gold trace) b, Diagram of PGH2 (gold) and AA (cyan) bound to the cyclooxygenase active site. Mobile side chains are colored gold and cyan to match their PGH 2 and AA bound positions, respectively.
Notice the hydrophobic nature of the amino acids surrounding the arachidonic acd. Note as well the kinks in the arachidonic acid associated with 4 all-cis double bonds in the fatty acid.
The reaction mechanism appears to involve a free radical. A possible free radical cascade mechanism, based on recent work by Marnett et al. and Kiefer et al. is shown and summarized below. Note that amino acids important for catalysis listed below do not appear be in the same positions as shown in the figure above. Mutagenesis clearly shows the importance of Tyr 385 in catalysis. The crystal structure above represented an alternative binding mode for arachidonic acid/COX2 that would not be catalytically active.
- The carboxylate of arachidonic acid is coordinated to Arg 120 and Tyr 355.
- The C13 pro(S) H atom of arachidonic acid is close to Tyr 385 which allows its abstraction.
- This results in a radical centered on C11 that reacts with dioxygen to form a peroxyl radical
- Attack by dioxgen at C11 occurs from the side of the substrate opposite to that of hydrogen abstraction
- The oxygen radical at C11 cyclizes by attacking C9.
The C13 proS hydrogen atom (not proton) is remove from bound arachidonic acid by a free radical form of Tyr 385, which acts as an oxidizing agent. A site-specific mutants in which Tyr 385 is replaced by Phe is inactive. How is the Tyr free radical formed? Based on single electron standard reduction potential (0.9 V for Tyr and -0.2 to + 0.2 V for Fe3+ in the bound heme), it appears that the heme iron is not a potent enough oxidizing agent to accomplish this task. However, oxygen bound to the heme iron could be converted to a peroxide and form an Fe4+-oxo complex much like we saw step 7 of a possible reaction pathways for cytoP450cam. The Fe4+ ion is a more potent oxidzing agent (standard reduction potential of approximately 1V, sufficient for oxidation of Tyr 385. Another possibility is that the peroxide activator (in the formation of the ferryl-oxo ligand) is NO (nitric oxide, a free radical). NO is formed by immune cells (like macrophages) on immune activation. The NO might react with superoxide (also a radical, possibly formed during an oxidative burst in macrophages during immune stimulation) to form peroxynitrite (NO3-). This can donate an oxo group to the Fe3+ to form the Fe4+-oxo complex, which could then oxidize Tyr to the free radical form. There might be other mechanisms as well to generate the Tyr free radical, since just adding organic peroxides to the enzyme will generate it.. After abstraction of the proS H atoms, a carbon-centered free radical at C11 results which reacts with oxygen as shown below. The exact form of oxygen that reacts is unclear, but presumably is either a peroxy or activated singlet form.
Figure: Prostatglandin Synthesis: Possible Mechanism
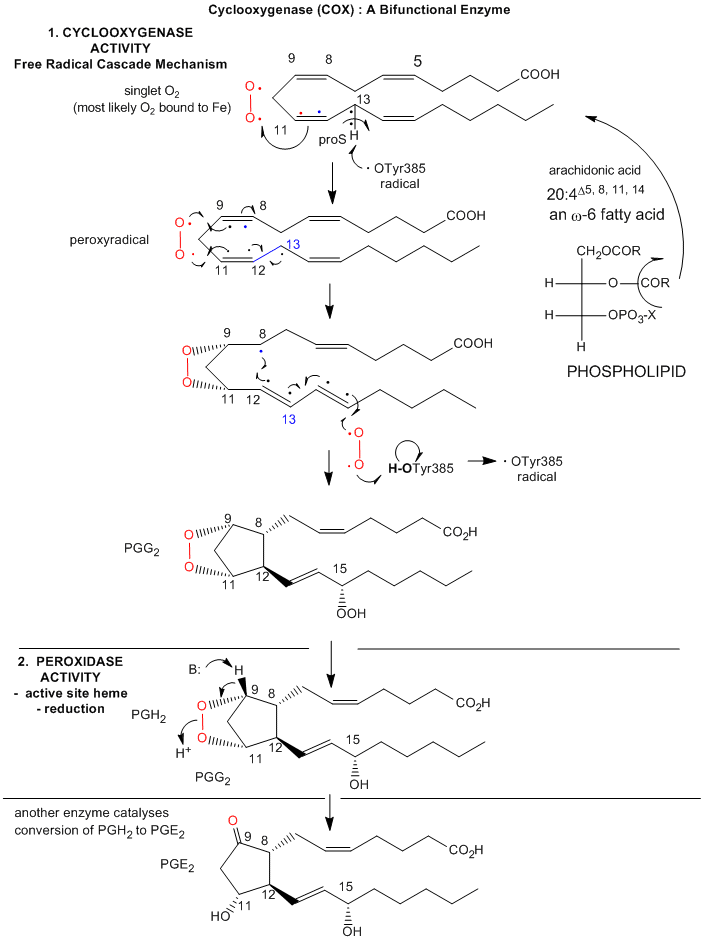
Prostaglandins, which were first isolated from prostate glands, serve as powerful, but labile local hormones which are mediators of pain, inflammation, immune and clotting activity. The cyclooxygenase activity is inhibited by aspirin, which probably accounts for most of its anti-inflammatory and analgesic properties. Aspirin, acetylsalicylic acid, acetylates a reactive Ser 530 in the active site. Another nonsteroidal anti-inflammatory drug (NSAID) with similar properties is Ibuprofen (Advil). Acetaminophen (Tylenol) is also considered a member of this drug class, even though it doesn't have anti-inflammatory properties. The question has arisen as to why. It now turns out that there are apparently three different types of COX, I, II, and III. COX III is expressed in the brain, and might be involved in pain pathways. Acetaminophen appears to work on this COX, as shown in the chart below (Bazan et al.).
Cyclooxygenase Activities
| COX | Expression | Function | Inhibitors |
| COX 1 | constitutively | organ pain, platelet function, stomach protection | NSAIDs including aspirin |
| COX 2 | induced by growth factors, neurotransmitters, inflammatory cytokines, oxidative stress, injury. Constitutively in brain, kidney | Inducible COX2: inflammation, pain, fever Constitutive COX2: synaptic plasticity |
NSAIDs, COX 2 inhibitors including celecoxib (Celobrex ) which has few GI problems associated with its use |
| COX 3 | constitutively, high in brain, heart | pain pathways, not inflammation pathways | acetaminophen (no GI problems, great fever reducer), some NSAIDs |
![]()
![]() Jmol:
Cyclooxygenase I and II
Jmol:
Cyclooxygenase I and II
Fish n-3 fatty acids and health
We mentioned the importance of arachidonic acid in signal transduction in the lipid chapter. In addition, the importance of n-3 fatty acids to health was discussed as well. As mentioned above, arachidonic acid is cleaved from the C2 or sn-2 position of membrane phospholipids and modified by cyclooxygenase or lipoxygenase to form prostaglandins and leukotrienes, both potent local biological mediators. Linoleic acid and 22:6n-3 (DHA or docosohexaenoic acid) are also found in membrane phospholipids at the sn-2 position. What is the mechanism for the health-protective effects of n-3 fatty acids like DHA?
In human tissue, DHA, 22:6n-3 or 22:6Δ5,8,11,14,17,20 is the most abundant n-3 polyunsaturated fatty acids (PUFAs). Since it is synthesized from linolenic acid (as is EPA), a deficiency of linolenic acid in the diet will lead to lowered levels of 22:6n-3 in tissues, with ensuing health effects. Since these lipids are involved in membrane structure, signal transduction, and hormone synthesis, diverse effects of dietary n-3 PUFA deficiency will be observed. 50% of all fatty acids in the sn1 and sn2 position of membrane phospholipids of rod outer segments (in the retina) are 22:6(n-3). Cognitive dysfunctions (loss of memory, etc.) have been linked to decreased levels of 22:6(n-3) in the brain. This fatty acid binds to retinoid X receptors which then activate (through linked binding reactions) nuclear receptors, leading to alterations in gene transcription in the CNS.
In other tissues, 22:6(n-3) rarely exceeds 10% of membrane fatty acids, but this percentage can be increased in cells with increases in a precursor, 20:5(n-3). DHA might affect lipid rafts in the membrane, which would affect movement of important membrane protein receptors (and associated proteins) in the membrane, altering cell response to environmental stimuli. DHA and EPA affect arachadonic acid conversion to prostaglandins and leukotrienes. EPA binds less tightly to cycloxygenase I and is a poor substrate for the enzyme, both effects which inhibit the formation of prostaglandins and signaling processes mediated by them. This explains why n-3 fatty acids have anti-inflammatory effects.
In addition, n-3 fatty acids have noticeable effects on gene transcription, which remain as long as these fatty acids are present in high levels in the diet These and other fatty acids bind to fatty acid-activated transcription factors called PPARs (peroxisome proliferator receptors - alpha, beta and gamma 1 and 2). These receptors regulate, through alterations in gene expression, proteins involved in lipid metabolism. Other fatty acid-dependent transcription factors are known as well. PPAR's bind 20:5(n-3) with a micromolar Kd and change the conformation of the protein to a form than can bind other proteins, ultimately altering gene expression.
Table: Biological Effects of n-3 polyunsaturated fatty acids EPA (20:5) and DHA (22:6)
| Organ(s) | Effect | Mechanism acts through |
| central nervous system | improve cognitive function | membrane composition; retinoic X receptor alpha |
| retina | improve acuity | membrane composition |
| immune | immunosuppressive; antinflamatory | membrane composition; rafts |
| cardiovascular | anti-arrhythmia; anti-clotting |
membrane composition; rafts; eicosanoids |
| serum lipids | lowers triglycerides (risk factor for cardiovas. dis) | peroxisome proliferator receptor alpha and gamma |
| liver | decrease lipid synthesis; increase fatty acid oxid. decrease VLDL synthesis |
sterol reg. element bind. protein; PPAR alpha PPAR alpha |
Adapted from Jump D. The Biochemistry of n-3 Polyunsaturated fatty acids. J. Biol. Chem. 277, pg 8755 (200)
B8. OXIDASES
This class of enzymes does not incorporate dioxygen into an organic substrate. Rather it accepts electrons released from an organic substrate, through intemediate electron carriers (such as ubiquinone and cytochrome C) to form superoxide (as in NADPH-oxidase), hydrogen peroxide (as in xanthine oxidase) or water (as in cytochrome C oxidase). The mechanism of cytochrome C oxidase again supports our expectations about enzymes that use dioxygen. Dioxygen binds metals in the enzyme. One oxygen atom binds a heme Fe2+ of cytochrome a3 which is bound to the enzyme, while the other binds a Cu1+ of Cu B. All oxygyen reduction intermediates remain bound to the enzyme. Four electrons are added from four different cytochrome C molecules, which serve as mobile carriers of electrons.
![]() Jmol: Updated
Cytochrome C Oxidase
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
Cytochrome C Oxidase
Jmol14 (Java) |
JSMol (HTML5)
Figure: Oxidases: Examples
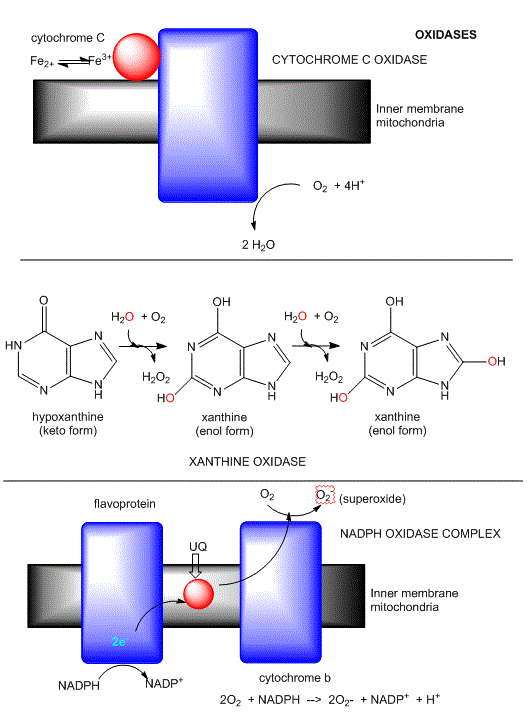
Another example of an oxidase is monoamine oxidase. Mitochondrial monoamine oxidase catalyzes the oxidative deamination of certain neurotransmitters after they have been taken up by post-synaptic neurons, in a process of inactivation. A reaction is shown below.
Figure:
Monoamine oxidases
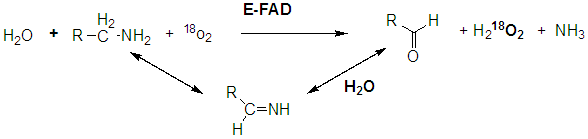
A Schiff base is formed which is then hydrolyzed, incorporating unlabeled oxygen into the oxidized molecule.
BIOLOGICAL OXIDATIONS: METHANE TO CO2
In the previous chapter section, we discussed the progressive oxidation of methane by 2 electron loses to form methanol, formaldehyde, formic acid and CO2, with a progressive increase in oxidation number for the C by +2 (from -4 in methane to +4 in CO2).
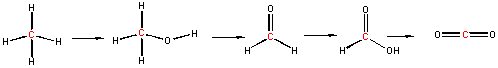
Methanotrophs are aerobic bacteria that use methane as a source of energy, converting it in a series of two electron oxidations as shown above, to carbon dioxide. The enzymes involved in this sequential process are methane monooxygenase, methanol dehydrogenase, formaldehyde dehydrogenase, and formate dehydrogenase. Methane monoxygenase exists in a soluble and membrane form, both of which are part of a larger complex. Both have a hydoxylase (which uses dioxygen to add O to methane) and the membrane form has recently been shown to be associated with have methanol dehydrogenase in a larger complex consisting of trimers of each enzyme (the hydroxylase and the dehydrogenase).
B9. Heme Proteins
So far in this course, we have examined three different kinds of heme proteins..
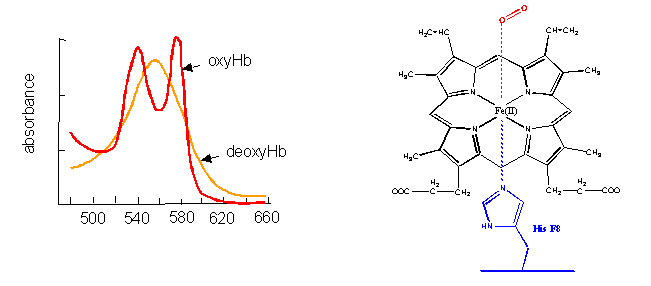
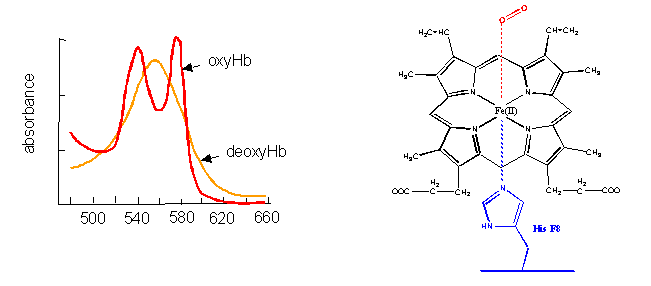
- The first, hemoglobin (and myoglobin) serve as carriers of dioxygen. Even though they bind one of the best oxidizing agents around (dioxygen), the heme Fe2+ does not get oxidized to Fe3+. If it does, as in the case of met-Hb, the protein looses it ability to carry oxygen.
Figure: hemoglobin (and myglobin) serve as carriers of dioxygen
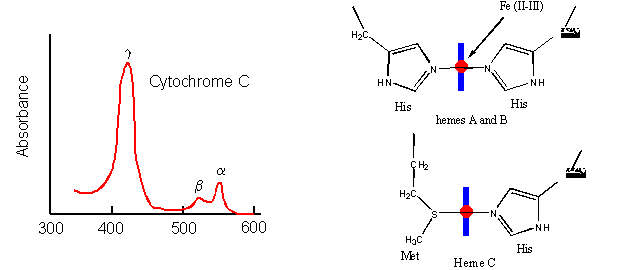
- Cytochrome C, on the other hand, does not bind dioxygen but rather serves as a carrier of electrons which get passed to dioxygen in Cytochrome C oxidase. Its Fe ion readily cycles between the 2+ and 3+ states as it serves as an electron carrier.
Figure: Cytochrome C
- Finally, the Fe2+ in the heme of the cytochrome P450s (so named since they have an absorbance maximum at 450 nm when they bind CO) does both. It binds dioxygen and cycles between the 2+ and 3+ states as it activates dixoxygen for hydroxylation reactions.
Figure: cytochrome P450s
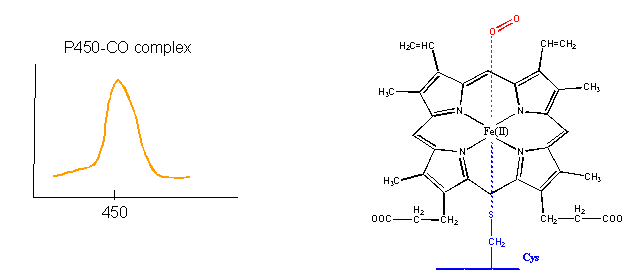
How could heme serve such diverse functions? We can explain this by referring to one of the main themes of the course - structure mediates function. The environment of each heme must be different. Clearly the protein ligands coordinating the Fe ions are different. The 5th ligand is the proximal His in hemoglobin while dioxygen binds to the 6th site. In cytochrome C, the 5th and 6th ligands are His and Met, respectively. In cytochrome P450, the 5th site is occupied by Cys, and the 6th by dioxygen. Presumably the environments surrounding the hemes are different as well. Once again, we have seen analogous example in which chemical properties are influenced by the microenvironment. The pKa of a given amino acid side chain can vary considerably depending on the polarity of the local environment. Likewise, the standard reduction potential of tightly bound FAD/FADH2 depends on the microenvironment.
As we have seen (from the study of heme proteins and the oxidative enzymes of cells), transition metals such as Fe, Zn, and Cu have vital biological roles as binding sites and cofactors in many reactions. Yet they also pose problems since they can lead to oxidative damage in cells. As we saw with cytoplasmic metallothioniens, which bind to heavy metals and protect the cell from such damage, many proteins are involved in binding and regulation of transition metals in the cell. Integral membrane proteins are required to bind and transport these cations into the cytoplasm. Other proteins act as sensors of transition ion concentration (such as latent transcription factors which bind heavy metals and become active transcription factors for metallothioniens. Others act as chaperone proteins which bind metal ions and transfers them to apometalloproteins. Recent work has suggested transporters and chaperones involved in metal ion biology bind these ions with unusual coordination geometry, which presumably facilitates transfer of the ion to the apo-target protein.
The transition metals Zn and Fe are often found in E. Coli at a concentration of 0.1 mM, compared to Cu and Mn which are present at concentrations from 10 to 100 μM. Also, about one third of all proteins demonstrate specific binding of metal ions and can be classified as metalloproteins. Mass balance suggests that metal ions would be distributed in proteins with low, intermediate, and high metal binding affinity as well as in free pools, which which potentially be toxic to cells. Metalloproteins, depending on their Kd for metal ion binding, would hence be in various state of ligation. The free concentration of some ions (Cu and Zn) is so low that newly synthesized apoproteins which bind these ions would not obtain the ion from the free pool. In such cases, metal chaperones would be required.
B10. Links and References
-
- Zhao et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 451, 587-590 (2008)
- Marie Lagouge, M., ...., Auwerx, J. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. 10.1016/j.cell.2006.11.013 (Nov 2006)
- Baur, J., ...., Sinclair, D. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337 - 342 (2006)
- Myronova, N. et al. Three-Dimensional Structure Determination of a Protein Supercomplex That Oxidizes Methane to Formaldehdye in Methylococcus capsulatus (Bath). Biochemistry, 45, 11905 (2006)
- Wood et al. Sirutin activators mimic caloric restriction and delay ageing in metazoans. Nature. 430, pg 686 (2004)
- Anderson, R. et al. Yeast life span extension by caloric restriction is independent of NAD fluctuation. Science. 302, pg 2124 (2003)
- Hydrogenases: Science, 299, pg 1686 (2003)
- Aguilaniu, H. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science, 299, pg 1751 (2003)
- Jump, D. The Biochemistry of n-3 polyunsaturated fatty acids. J. Biol. Chem. 277, pg 8755 (2002)
- Finney, L. and O'Halloran, T. Transition Metal Speciation in the Cell: Insights from the Chemistry of Metal Ion Receptors. Science, 300, pg 931 (2003).
- Ohtake et al. Modulation of estrogen receptor signaling by association with the activated dioxin receptor. Nature 423, pg 487, 545 (2003)
- Bazan, N. G. and Flower, R.J. Lipid Signals in Pain Control. Nature. 420, pg 135 (2002)
- Kiefer et al. Structural Insights into the stereochemistry of the cyclooxgenase reaction. Nature. 405, pg 97 (2000)
- Marnett et al., Arachidonic Acid Oxygenation by COX-1 and COX-2. J. Biol. Chem. 274, pg 22903 (1999)
- Oxygenase Pathways: Oxo, Peroxo, and Superoxo. Science. 292, pg 651 (2001)
- Malkowski et al. The Productive Conformation of Arachidonic Acid Bound to Prostaglandin Synthase. Science. 289. pg 1933 (2000)
- Lin et al. Requirement of NAD and SIR2 for Life-Span Extension by Calorie Restriction in S. Cerevisiae. (role of NAD) Science. 289. pg 2062, 2126 (2000)
- Imai, S. et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature, 403, 795 (2000)
- Landry, et. al. The silencing protein Sir2 and its homologs are NAD-dependent protein deacetylases. PNAS, 97, pp 5807 (2000)
- A radical approach to (cancer) treatment (reactive O2 species). Nature. 407, pg 309, 390 (2000)
- Melov et al. Extension of LIfe-Span with Superoxide Dismutase/Catalase Mimetics. Science. 289. pg 1567 (2000)
- Anti-inflammatories inhibit cancer growth - but how? (about cyclooxygenase I and II) Science. 291. pg 581 (2001)
- Schlicthling et al. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 287, pg 16 (2000)

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.