Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
H: PROTEIN AGGREGATES AND DISEASE
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 03/10/16
|
Learning Goals/Objectives for Chapter 2H: After class and this reading, students will be able to
|
H1. Protein Aggregation
We have seen that protein aggregates complicate the lives of people who study protein folding in vitro and who try to express human proteins in prokaryotes like E. Coli in vivo. Instead of viewing these aggregates as junk, some now study them avidly. It turns out that these aggregates are not as non-specific as earlier believed. In addition, an understanding of how and when they form will give us clues into the etiology and treatment of some of the most debilitating and feared diseases. Much of this review is based on the following reference: Taubes, G. Misfolding the Way to Disease, G. Science, 271, 1493-1495 (1996)
Clues Showing the Specificity of Aggregate Formation
- 1970's: It was shown that chymotrypsinogen could not be folded in vitro without aggregates forming. An intermediate was presumed to have formed that if present in high concentration would aggregate irreversibly instead of fold to the native state. Refolding of tryptophanase showed that it aggregated only with itself, suggesting specificity.
- 1981: King, at MIT, found a single amino acid folding mutant in a viral protein. Both the normal and mutant viral protein unfold at high temperature, but only the mutant would aggregate at high temperature, suggesting that aggregation could be programmed into or out of a gene
- mid 80's: The biotech industry, struggling to express growth hormone, found that a single amino acid change in bovine growth hormone completely prevented aggregation without affecting correct folding.
This knowledge of protein folding and aggregation was soon turn toward understanding several diseases in which protein aggregates were observed which either initiated or were associated with disease. These protein aggregates were termed "amyloid deposits" and seemed to be associated and perhaps causative of several neurodegenerative diseases. The name amyloid was first used by a German pathologist, Rudolf Virchow, who in 1853 described waxy tissue deposits associated with eosinophils (a type of immune cell). These deposits seemed to resemble starch (made of amylose and amylopectin) so he termed them amyloid. All known amyloid deposits are, however, composed of protein, not starch. It now appears that these disease may be caused by improper protein folding and subsequent aggregation. Except in certain rare inherited diseases, the amyloid deposits are composed of normal proteins (often called wild-type proteins) which seems to polymerize into fibrils. Sometimes, in inherited conditions, or when mutations appear in a specific protein, the amyloid protein deposits consist of the mutant protein. The proteins in these deposited fibers are composed predominantly of beta sheets which are perpendicular to the fiber axis, while the polymerized protein usually has little beta sheet structure. Examples are given below:
- Familial amyloidotic polyneuropathy (FAP) - Affects 1/10,00 to
1/100,000 people. The protein involved is called transthyrein, which
normally exists in blood as a tetramer formed by association of 4
identical monomers. In mildly acid condition in vitro, the equilibrium
between tetramer and monomer is shifted to monomer, which can aggregate
into fibrils. This aggregation could be promoted by possible transition
to a molten globule (discussed previously with lactalbumin) like state.
This has secondary structure but loosely packed tertiary structure with
more exposed hydrophobes. If the concentration is high enough the molten
globules aggregate. In people with the disease, mutations in the protein
destabilize the tetramer, pushing the equilibrium to the monomer, which
presumably increases molten globule formation and aggregation.
Specifically, Val30Met and Leu55Pro mutations promote dissociation of
the tetramer and formation of aggregates. Conversely, Thr119Met
inhibits tetramer dissociation. The aggregates deposit in heart, lungs,
kidney, etc, leading to death.
- Light Chain Amyloidosis; Light Chain Deposition Disease - The light
chain protein is a normal component of circulating antibody molecules.
Mutants in the light chain cause a destabilization of the native state
to state similar to a molten globule, which then aggregates.
- Lysozyme amyloidosis - This protein, with extensive alpha-helix
structure, is usually involved in carbohydrate catabolism. Two mutants,
Asp67His and another, Ile56Thr (normal amino acid/number in
sequence/mutant amino acid) destabliize the protein structure (as
indicated by a decrease in the Tm of about 10 degrees C) to a
molten-globule form, which probably aggregates to fibrils characterized
by extensive beta structure.
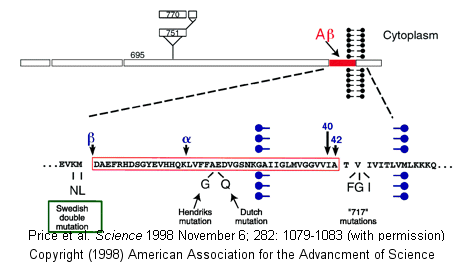
- Alzheimers-This disease involves a defect in a protein normally found in the membrane of neurons. The protein, called beta-amyloid precursor protein (BAPP), is a transmembrane protein. A slightly truncated, soluble form is also found secreted from cells and is found in extracellular fluid (such as cerebrospinal fluid and blood). The normal function of these BAPP proteins is not yet clear. An endoprotease cleaves a small 40-42 amino acid fragment from this protein, forming the amyloid beta (Ab) protein. It is this protein or a mutant form of it which aggregates to form beta-sheet containing fibrils in Alzheimer's disease. Several mutations in different proteins have been linked to Alzheimer's, but they all seem to increase production or deposition or both of the amyloid beta protein. These deposited plaques are extracellular, and have been shown to cause neuronal damage. The are found in areas of the brain required for memory and cognition. The BAPP gene is found on chromosome 21, the same chromosome which is present in an extra copy (trisomy 21) in Downs Syndrome, whose symptoms include presenile dementia and amyloid plaques. Aggregate formation appears to be driven by increased expression of BAPP and hence amyloid beta protein. In addition, some mutants may serve to destabilize the amyloid beta protein, increasing its aggregation.
- Transmissible spongiform encephalopathies (TSEs) - Including scrapie in sheep, bovine spongiform encephalopathy (mad cow disease), and in humans Creutzfeld-Jacob Disease (CJD), Fatal Familial Insomnia (FFI),Gerstman-Straussler-Scheinker Syndrome, and Kuru (associated with cannibalism). In these fatal diseases, the brain, on autopsy, resembles a sponge with holes. In contrast to the diseases above, these diseases can be transmitted from one animal to another, but typically not between species. (However, consider the controversy with mad cow disease.) Also, the infectious agent can self-replicate in vivo. The logical conclusion is that a virus (slow-acting) is the causative agent. However, the infectious agent survives radiation, heat, chemical agents, and enzymes designed to kill viruses and their associated nucleic acids. Mathematical analyses suggested that the infectious agent in such diseases could be nothing more than a protein. Stanley B. Prusiner in the 80's isolated just such a protein which he named a prion, for proteinaceous infectious agent. Since then he and others have amassed substantial evidence to support his contention. In October 1997 he was awarded the Nobel Prize in Medicine.
 Navigation
Navigation
Return to Chapter 2H: Protein Aggregates and Disease
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2H: Protein Aggregates and Disease

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.