Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
H: PROTEIN AGGREGATES AND DISEASE
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 03/10/16
|
Learning Goals/Objectives for Chapter 2H: After class and this reading, students will be able to
|
H2. Prions and Disease
The normal cellular form of this protein, PrPc is highly conserved in mammals, and is widely expressed in embryogenesis. Techniques exist to delete or make ineffectual genes in mice. When a mice knockout of the PrPc (i.e. the gene for the protein was deleted in all cells) was made, the mice appeared normal. More recent data suggests however, that these mice had altered circadian rhythms and sleep patterns, which suggest a possible link to Fatal Familial Insomnia. The PrPc is a normal membrane protein in neurons. It is anchored to the membrane through a glycosyl-phosphatidyl inositol link, with the protein chain on the outside of the neuronal plasma membrane. The PrPc (without the PI link) is water soluble, protease sensitive, and consists of 42% alpha helix and 3% beta sheet.
![]() Jmol:
Updated Prion Protein, Mad Cow Disease, and Mutations
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Prion Protein, Mad Cow Disease, and Mutations
Jmol14 (Java) |
JSMol (HTML5)
The problem in the transmissible spongiform encephalopathies (TSE's) is that amyloid-like protein aggregates form which appear to be neurotoxic. The protein found in the plaques (in cases other that those that are inherited) has the same primary sequence as the PrPc but a different secondary and presumably tertiary structure. The protein found in the plaques, called the PrPsc (the scrapie form of the the normal protein) is insoluble in aqueuous solution, protease resistant, and has a high beta sheet content (43%) and lower alpha helix content (30%) than the normal version of the protein PrPc.
Figure: Cartoon Models of PrPc and PrPsc
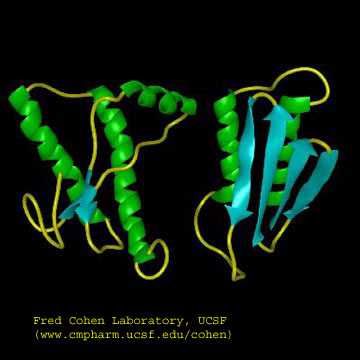
A genetic, inheritable form of disease also exists, in which a mutant form of the PrPc occurs, whose normal structure is destabilized by the mutation. The aggregates caused by the mutant form of the disease are understandable in light of the other diseases which we discussed above. The question is how does the normal PrPc form PrPsc . Evidence shows that if radiolabeled PrP*c from scrapie free cells is added to unlabeled PrPsc from scapie infected cells, the PrP*c is converted to PrP*sc! It appears that the PrPc protein has two forms not that much different in energy, one composed of mostly alpha helix and the other of beta sheet. A dimer of PrPc.PrPsc might form, which destabilizes the PrP*c causing a conformational shift to the PrPsc form, which would then aggregate. Exposure to the PrPsc form would then catalyze the conversion of normal PrPc to PrPsc . Hence, it would be transmissible by contact with just the PrPsc form of the protein. Likewise species specificity could be explained if only dimers of PrPc.PrPsc formed from proteins of the same species could occur. The inherited form of the disease would be explained since the mutant form of the normal protein would more easily form the beta structure found in the aggregate.
It has recently been found that the very same mutation in PrPc, Asp178Asn can cause two different diseases - CJD and FFI. Which disease you get depends on if you have 1 of two naturally occurring, nonharmful variants at amino acid 129 of the normal PrPc gene. If you have a Met at that position, and acquire the Asp178Asn mutation, you get CJD. If, on the other hand, you have a Val at amino acid 129 and acquire the Asp178Asn mutation, you get FFI. This disease was first observed in 1986 and has been reported in five families in the world. It occurs in the late 50's, equally in men and women. It is characterized by a progressive loss of the ability to sleep and disrupted circadian rhythms. The brain shows neuronal loss. It is known that amino acids 129 and 178 occur at the start of alpha helices, as predicted from propensity calculations. Chronic exposure to micromolar levels of synthetic fragment 106-126 of PrPc kills hippocampal neurons. This peptide also has the greatest tendency to aggregate synthetic PrPc peptides.
A series of recent studies have expanded on our knowledge of prion structure. Nelson et al. have obtained the crystal structure of a fibril aggregate made of a short peptide (7 amino acids) from the yeast prion protein Sup35. As presumably occurs in amyloid fibers, these crystals show beta-sheet structures stacked vertically to produce fibril structures. The unit of stacking appears to be pairs of beta-sheets, with the inner side amino acids of one member of the pair interacting with the inner amino acid side chains of the other member of the pair, in a process which excludes water. Similar studies by Ritter et al, using NMR and fluorescence, found pair of beta sheets to be the motif of the fibril. Using fluorescence, they identified two regions, each 15 amino acids, important in collapse to molten-globule like state for nucleation of fibril formation.
Kuru killed many members of the Fore tribe in New Guinea until the cannibalistic practice of eating dead relatives was stopped. Analysis of the genes for the prion protein in the Fore tribe and other ethnic groups in the world show two version differing by just one amino acid in all people (remember that a single gene is represented in both maternal and paternal chromosomes. That these two forms exist through the world suggest that they have been selected for by evolution and confer some biological advantage. People who have just one form of the protein are more susceptible to the development of prion diseases. Mead and Collinge have shown that about 75% of older Fore women (who had lived through cannibalistic practices) had two different prion genes, compared to about 15% of women from other ethnic groups. This high percentage suggests that these women were protected from the disease, leading through natural selection to a high percentage of heterozygotes in this defined population. The general presence of two forms of the prion gene (which probably offers protection from prion disease) suggests that cannibalism might have been widespread in our early ancestors.
There appears to be one main difference between the formation of amyloid fibers from prion proteins and others such as mutant lysozymes. If you add mutant lysozyme to normal lysozyme, the amyloid fibers contain only the mutant protein. However, if you incubate mutant prion proteins with normal prions, the normal proteins become pathological.
QED - Protein Aggregates are not just test tube artifacts, but rather matters of life and death.
![]() CDC:
Prion Disease
CDC:
Prion Disease
![]() Jsmol:
Jsmol:
![]() Protopedia
- Prions
Protopedia
- Prions
 Navigation
Navigation
Return to Chapter 2H: Protein Aggregates and Disease
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2H: Protein Aggregates and Disease

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.