Biochemistry Online: An Approach Based on Chemical Logic

MOLECULAR MECHANICS AND DYNAMICS
04/13/16
C. Molecular Mechanics
The following review is based on an NIH Guide to Molecular Modeling, which was removed from the web, to the best of my knowledge, in 1996. I've recreated many of the graphs using Excel.
Molecular mechanics uses Newtonian mechanics to calculate energies of atoms in large molecules like proteins. It assumes that nuclei and electrons are one particle with radii and calculated charged. Bonds are treated as springs connecting atoms. Energies are calculated classically (not with quantum mechanics). Parameters, many based on QM calculations on small molecules, are assigned to all bonds, angles, dihedrals, etc. Interactions are bonded (local) and nonbonded.
Bonded interactions involved atoms connected by one bond (bond stretch), two bonds (angle bending) and 3 bonds (dihedral angle change).
.
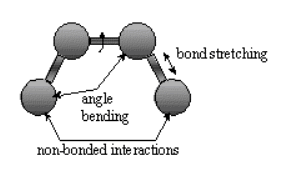
Non-bonded atoms (greater than two bonds apart) interact through van der Waals attraction, steric repulsion and electrostatic attraction/repulsion.
All energy terms from these interactions are summed to give the energy of a given conformation. The energy should be considered relative to those of other conformations. Here is the basic energy equation for all of these energy terms:
Energy (E) = E Stretch + EBending + ETorsion + E Non-bonded Interactions
The "force field" consist of the energy equations and the parameters for each of the energy terms. There are many different commercially available force fields.
 Navigation
Navigation
Return to Molecular Mechanics and Dynamics Contents
Return to Biochemistry Online Table of Contents
Archived version: Molecular Mechanics and Dynamics

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.