Biochemistry Online: An Approach Based on Chemical Logic

MOLECULAR MECHANICS AND DYNAMICS
04/13/16
F. Summary Interactions
Some programs assign charges using rules or templates, especially for macromolecules. In some force-fields, the torsional potential is calibrated to a particular charge calculation method (rarely made known to the user). Use of a different method can invalidated the force-field consistency. Sometimes, an additional bonded interaction term, improper dihedrals, are added as illustrated below. The potential for that is given by the following equation:
Eimproper = Σangles kω (ω - ωo)2
All of the potential energy functions are illustrated in the graph below, where V is the potential energy.
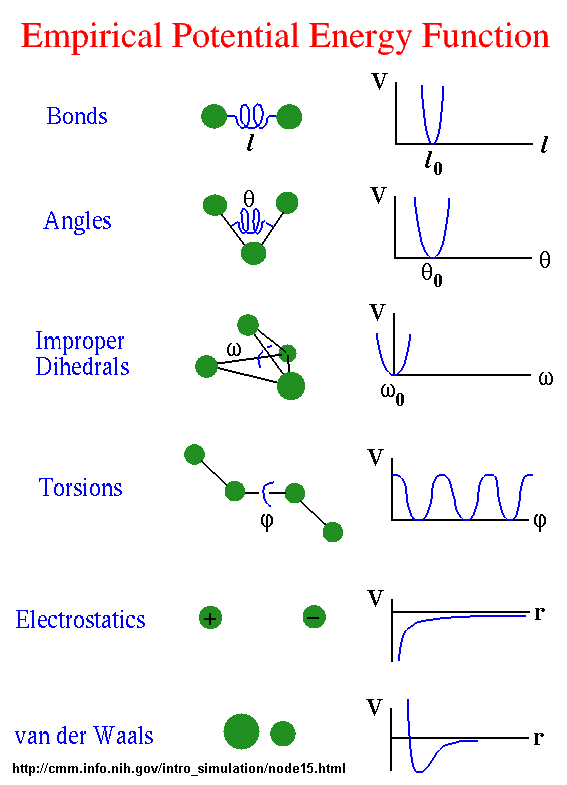
Go to this great web site by Sharon Hammes-Schiffer, Shaffer Associate Professor of Chemistry Theoretical and Computational Chemistry at Penn State to see interactive graphs for these potential functions.
 Navigation
Navigation
Return to Molecular Mechanics and Dynamics Contents
Return to Biochemistry Online Table of Contents
Archived version: Molecular Mechanics and Dynamics

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.