Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 9 - SIGNAL TRANSDUCTION
D: mTOR and Nutrient Signaling
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 6/22/16
|
Learning Goals/Objectives for Chapter 9D:
|
D6. Regulation of mTORC1 by Insulin and Growth Factors
mTORC1 is clearly regulated by local factors (amino acids, energy state) and systems factors (growth factors). This list is growing daily. The following have been shown to lead to mTORC1 activation including small molecules such as amino acids, ATP (through AMP kinase), oxygen and glucose, and larger ones such as insulin, other growth factors, cytokines (immune growth factors and regulators), oncogenes (which obviously promote cell proliferation) and some infectious agents. Other molecules or processes inhibit mTORC1, including tumor suppressors and stress.
mTORC1 promotes mRNA and protein synthesis as described above but also nucleotide and lipid synthesis, which is not described in detail above. In addition, it promotes aerobic glycolysis (Warburg effect), to supply not energy but intermediates for biosynthesis, as well as the pentose pathway, which forms NADPH for reductive biosynthesis and ribose for nucleic acid synthesis.
Lets look at 2 specific external hormones, insulin and epidermal growth factor (EGF), and how they affect mTORC1 activity;
Insulin:
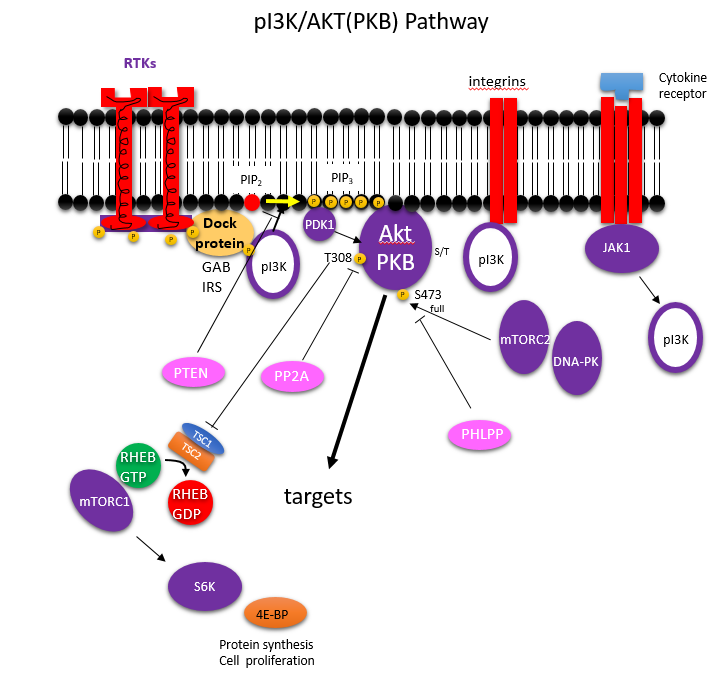
Insulin binding to its receptor leads to the activation through phosphorylation of the kinase Akt (aka Protein Kinase B) after upstream phosphorylation of membrane phosphoinositides in the membrane and activation of phosphoinositide-dependent kinase 1, PDK1. Atk, as shown in the signaling figure for AMPK, phosphorylates TSC2, the GAP for Rheb. The arrows on the AMPK kinase figure above is not consistent with our previous use of arrows. In the figure from CST, arrows show that both AMPK and Akt phosphorylate TSC2. The phosphorylated TSC is shown inhibiting Rheb, the small G protein. This would not make physiological sense. Phosphorylation of TSC2 by AMPK (signaling energy depletion) activates the TSC2 GAP protein which would inhibit RheB (the G protein) and hence inhibit mTORC1. In contrast, phosphorylation of TSC2 by Akt (signaling the abundance of glucose) leads to the inhibition of the GAP activity of TSC2. That would keep Rheb in the active, GTP bound form, which leads to the activation of the bound mTORC1. A more complete description of the pathway where insulin binds to its receptor (an insulin-gated receptor tyrosine kinase) and leads to activation of mTORC1 through Akt is shown below.
EGF:
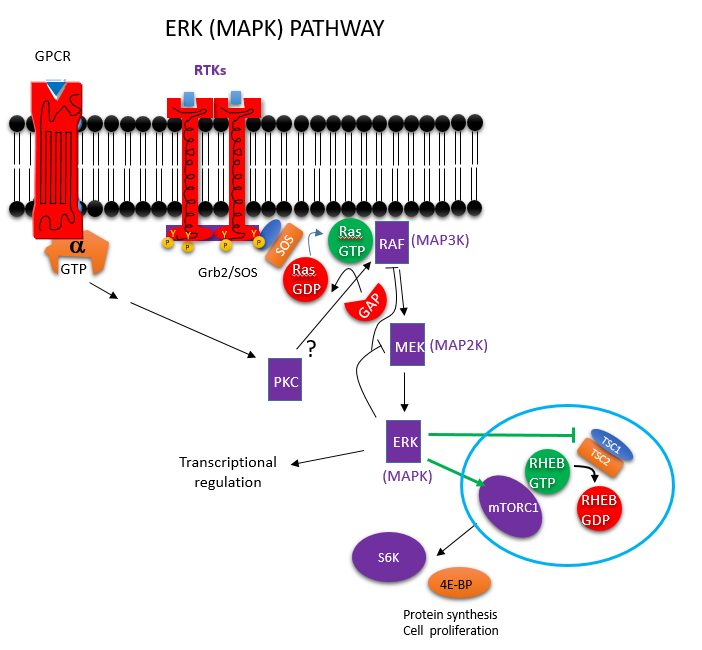
EGF binds its receptor, activating it as a receptor tyrosine kinase. Typical of other receptor kinases, it activates the mitogen activate protein kinase system. This process is mediated by Ras (a small G protein) activator of Raf (a mitogen activated protein kinase kinase kinase or MAP3K). Active Raf phosphorylates and activates MEK (a MAPK2) which activates ERK (a MAPK). Erk phosphorylates mTORC1 directly, which activates it. It also phosphorylates TSC2/TSC1, which inhibits this GAP proteins, leading indirectly to the activation or the small G protein Rheb, which also activates mTORC1. These steps are shown in the figure below.
A summary showing the kinases that activate or inhibit TSC2/1 is shown below. We tend to concentrate on our favorite protein and confer on it special status as critically important in a pathway. One could clearly pick the GAP protein TSC2 as especially important in regulating the activity of mTORC1, as shown below.
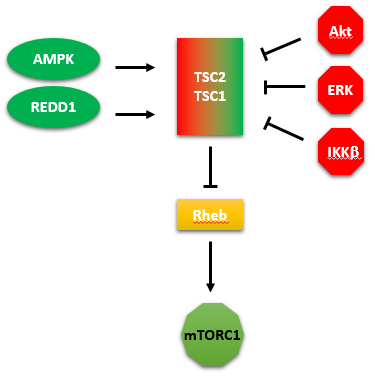
The figure above shows two additional proteins. One is REDD1 (not a kinase), which activates the GAP protein TSC2, leading to the inhibition of the small G protein Rheb, and hence the inhibition of mTORC1.
REDD1 (regulated in development and DNA damage responses 1) is also called DDIT4 (DNA-Damage-Inducible Transcript 4). It is a gene whose expression is activated during hypoxia by hypoxia-inducible factor-1 and also during DNA damage. Hypoxia obviously alters metabolism very quickly. The protein is degraded by the proteasome after it is targeted for degradation by post-translational addition of ubiquitin. This suggest yet another way to regulate the activity of mTORC1.
The other is IKK beta, also known as IKBKB ( Inhibitor Of Kappa Light Polypeptide Gene Enhancer In B-Cells, Kinase Beta). It is a kinase that phosphorylates and inhibits TSC2 that inhibits Rheb, leading to the activation of mTORC1.
This kinase is activated by many stimuli including inflammation (mediated by cytokines), bacterial or viral infections, and DNA damage. It phosphorylates a bound inhibitor of NF-kappa beta. This allows ubiquitinylation of the inhibitor, targeting it for proteasomal degradation. The free NFKB can then enter the nuclease and alter transcription of genes involved in the immune response and hence promote proliferation. Under these conditions, one would expect the activation of mTORC1.
A final note: Less is known about how lipids regulate mTORC1. Two possible lipid signaling molecules, phosphatidic acid and phosphatidyl inositol -3-phosphate are probably involved. The enzymse that make them, phospholipase D and phosphoinositide 3-kinase (from the PIK3C3 gene), also known as VPS34 (for vacuolar protein sorting from yeast), are found in phagosomal and lysosomal vesicles and are involved in their processing, seem to be involved in mTOR signaling. Obesity and people with high fat diets have elevated mTOR activity.
 Navigation
Navigation
Return to Chapter 9D. Nutrient Signaling Sections
Return to Biochemistry Online Table of Contents

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.