Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 5 - BINDING
E: MODERN METHODS IN DRUG DEVELOPMENT
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Updated: 03/31/16
|
Learning Goals/Objectives for Chapter 5E: After class and this reading, students will be able to
|
The difficulty lies, not in the new ideas, but in escaping the old ones, which ramify, for those brought up as most of us have been, into every corner of our minds. - John Maynard Keynes
E1. Introduction to Drug Development
Typically, drugs have been found by an almost serendipitous process. Active molecules are usually derived from biological sources such as bacteria, fungi, plants, etc. Different molecular species are extracted which, in bioassays, seem to inhibit a target biological function or specific enzyme. The structure is then solved, and medicinal chemists "decorate" the molecule by covalently modifying the initial structure in ways which will improve its medicinal value (decreased Kd, increased solubility, longer half-life, fewer side effects, etc.) Bioactive structures are often found to be peptides or modified peptides. One main job of a medicinal chemists is to synthesize a peptidomimetic with different functional groups than are found in peptides, making the structure more stable to endogenous proteases. Newer methods involve molecular modeling and rational drug design. Computers can be used to analyze the structure and activity of a large number of different molecules which inhibit the target enzyme. By doing Quantitative Structure Activity Result studies (QSAR), the computer can be used to hypothesize the structure of the "perfect" inhibitor, which can then be synthesized. In an analogous fashion, the active site of the target enzyme can be constructed from this data, without the benefit of having a crystal and NMR structure.
Drugs can be designed to inhibit enzymes or to bind to sites on receptors (either in the membrane or the cytoplasm). When bound to a receptor, the drug ligand may mimic the natural ligand and lead to expression of the biological activity of the receptor. In this case, the drug is an called an agonist of the normal ligand. In general, if the drug inhibits the activity of the bound receptor, the drug is called an antagonist. Examples are show below. Notice the similarity and differences in structures between the normal ligand and the drug. A more detailed description of agonist and antagonist will be present later in the discussion of enzyme inhibitors.
Figure: Receptor agonists/antagonist I
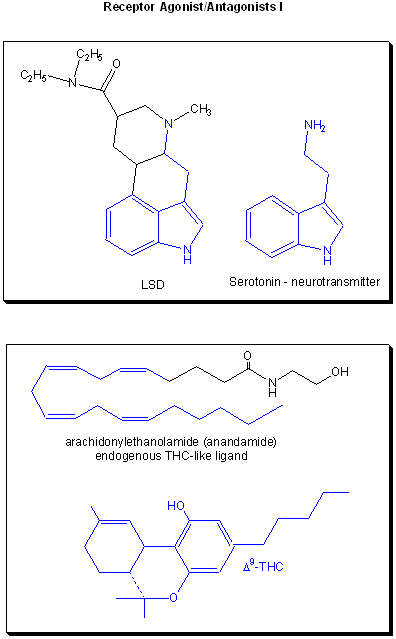
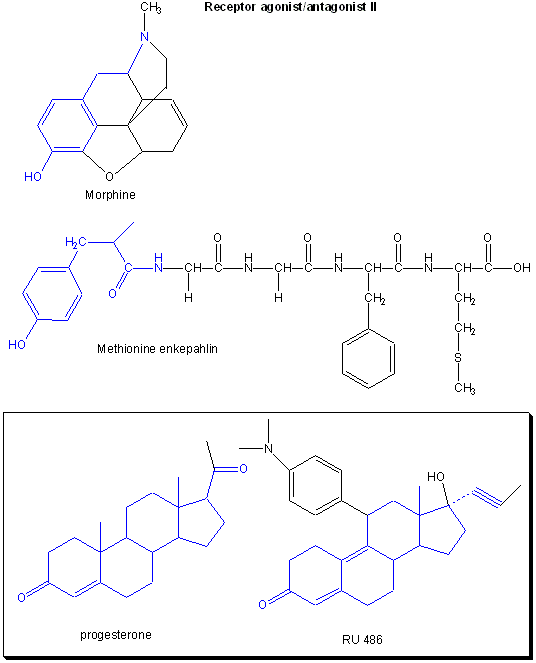
Figure: Receptor agonists/antagonist II
E2. Combinatorial Drug Development
Instead of searching various organisms to find a possible drug from all the extractable molecules of the organism, people are now synthesizing an enormous number of randomly constructed molecules, and then selecting active molecules from this random pool by binding or other bioassays. Solid phase synthesis of random peptides or nucleic acids can be used to generate thousands of possible test inhibitors. Those peptides or oligonucleotides that bind flurorescent-labeled protein can be determined, as shown below.
Figure: Combinatorial Drug Development.
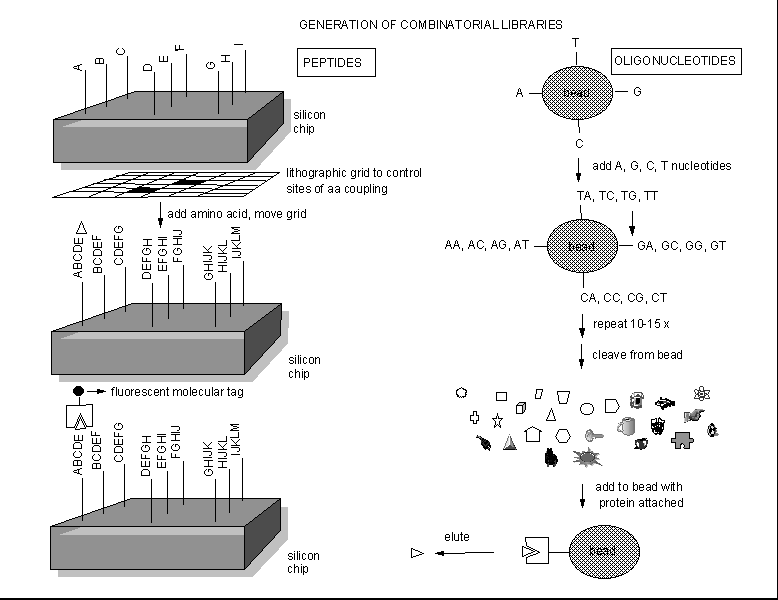
These structures can then be decorated or used to design peptide or oligonucleotide mimetics, which would be more resistant to enzymatic degradation. Another way to make a large library of peptide inhibitors is to make them using genetic engineering.
Recently, peptides made of D-amino acids have been synthesized. These are much more resistant to proteases than normal L-amino acid peptides. D amino acid peptides can only be made in the lab, and not through genetic engineering. An intriguing variation of this technique has recently been developed. Investigators have synthesized entire proteins in the lab using D-amino acids. Combinatorial L-peptide libraries were made using genetic engineering in bacteria. L-peptides were selected that bound with high affinity to the D-protein. The corresponding D-peptides were made in the lab, which then were found to bind to the normal L-protein.
In another combinatorial technique, single stranded RNA molecules called aptamers can be synthesized and tested for the binding/inhibitory activity for a enzyme. RNA molecules, which can form complex secondary and tertiary structure, can also present, given the appropriate sequence, a complementary binding surface to sites on proteins. Aptamers with high affinity are sequenced. Bases that appear to be necessary for high affinity binding are identified by comparing the sequence of different aptamers. This knowledge is then used to synthesize even tighter binding aptamers, in an process that mimics evolutionary selection for high affinity binding. Such a high affinity aptamer was recently made to bind to and inhibit Factor IXa, an active enzyme required for initiation and propagation of blood clotting. Traditional anticoagulant drugs are extremely valuable in treating and preventing inappropriate clot formation, which can lead to strokes (brain attacks) and heart attacks. The problem with these drugs is that they must not tip the clotting/anticlotting balance to far in the direction of clot prevention, since there are many times when clot formation is the appropriate biological response (such as in the prevention of hemorrhagic stoke). Rusconi et al. realized that once they had developed the high affinity aptamer for factor IXa binding, they immediately could synthesize an anedote for the aptamer. It would have a complementary sequence to the Factor IXa binding bases of the drug aptamer, that would allow it to bind to the original apatmer and form a double-stranded RNA sequence. The crystal structure of the homodimer transcription factor, NF-κB (p50)2 bound to an RNA aptamer has been determined. The RNA sequence is dissimilar to the DNA sequence which binds the transcription factor, even though both have similar dissociation constants.
![]() Jmol:
Updated Nf-Kb(P50)2
Complexed To A High- Affinity RNA Aptamer
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Nf-Kb(P50)2
Complexed To A High- Affinity RNA Aptamer
Jmol14 (Java) |
JSMol (HTML5)
E3. Computer Design of Drugs
Given a 3D structure of a protein, a clever synthetic chemist should be able to design a drug to fit into any "pit or canyon" that serves as a binding site for a biological ligand. Virtual reality simulations showing the protein target and drug with feedback on binding interactions provided to hand inputs using a haptic device are being used in drug design.
Many computer programs exists as well that will "dock" small ligand to active sites on large biological molecules.
-
 AUTODOCK:
Automatic Docking of Flexible Ligands to Macromolecules
AUTODOCK:
Automatic Docking of Flexible Ligands to Macromolecules -
Zinc:
A Free Digital Molecular Library Database for structure-matching
Drugs are intended to be target-specific; however, many drugs interact with other physiological targets which may result in side effects. In order to understand drug-target associations and predict new targets for established drugs, Keiser M. et. all used a similarity ensemble approach (SEA) to compare 3,665 FDA approved drugs to hundreds of different ligands. In this approach, they compared targets based on the similarity of ligands that bind to the different targets. This comparison predicted 257 drug-target associations that were not noted in either the MDL Drug Data Report or the World of Molecular Bioactivity database. Of these, they then tested 30 of the predictions. 14 of the 30 were found to have 23 previously unknown targets.
Keiser, et.al then went on to analyze if these new targets were biologically relevant. To do so, they used the following criteria:
- Did the new targets contribute to the primary activity of the drug,
- Did they cause side effects, or
- Were the new targets unrelated by sequence, structure, and function to the typical target.
When testing whether some of the new targets were primary sites of action, they noted that for some of the different drugs, the binding affinity was higher for the new target than it was for the intended target. For example, Keiser et. all used SEA to compile different ligands for dimethyltryptamine (DMT). Based on this, and DMT's side effects, they tested the binding affinity of DMT to different receptors and found that DMT would bind five of these. All of these receptors had substantially greater affinities for DMT than the intended target. Some of the new off-targets also gave insight into several drugs� side effects. Finally, there were four drugs that did not bind to targets that were related by sequence or structure to their intended targets. However, these new targets were related to the intended targets in that they had similar ligands regulating their activities. Overall, Keiser et. all were able to use the similarity ensemble approach based on ligand similarities to map new off-target associations for drugs. This approach can be used to gain better understanding of drug associations and to predict new molecular targets
E4. In Situ Click Chemistry in Drug Synthesis
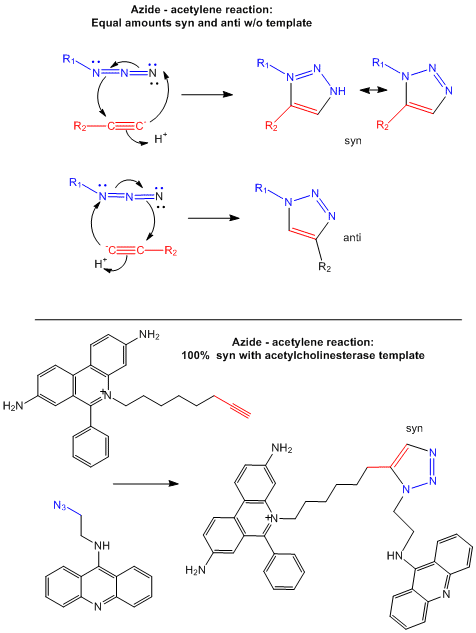
Drugs that inhibit enzymes typically bind to the active site of the enzyme where catalysis occurs. Binding of an inhibitor precludes binding of the normal reactants (substrates) for the enzyme, inhibiting its activity. In a new strategy, two small reactive molecules selected to bind independently in the active site can covalently react with each other to form a new drug with very high specificity and very high binding affinity (low Kd). Recently this has been used to synthesize noncovalent inhibitors of the enzyme acetylcholinesterase (Barry Sharpless Lab, Scripps Lab). The reactive groups chosen in the example below are azide and acetylene derivatives, which when held in close approximation in the binding site of the enzyme undergo a cycloaddition reaction to form a triazole.
The actual mechanism (not the simplified version show below) requires catalysis by cupper ions which forms a complex with the acteylide (deprotonated acetylene) effectively decreasing the pKa of the acetylene functional group, making it a better nucleophile. A dicopper intermediate is suggested in which the azide interacts with the second copper. Subsequen rearrangment lead to the triazole products.
Figure: Test tube and in situ synthesis of triazole inhibitors.
E5. DNA Binding Drugs
Most drugs in use today bind to specific proteins and interfere with the activity of the protein. Some antibiotics do bind DNA by intercholating nonspecifically between base pairs and interfering with replication and transcription. An alternative new approach is to design drugs that bind to specific sequences of DNA and either promote or inhibit the transcription of key genes. β-hydroxybutyrate is an experimental drug now being used to treat sickle-cell anemia. Somehow it increases the transcription of fetal hemoglobin genes, which form proteins which do not aggregate as the sickle cell hemoglobin gene product does. These approach has been called chemical genetics. Stuart Schreiber at Harvard has been a leader in the this field.
-
Article
on Schreiber's work
-
Schreiber's
company, Infinity Pharmaceuticals
Another way to inhibitor specific gene transcription is to bind a small single-stranded DNA to the dsDNA to form a triple helix. The single stranded DNA molecule can bind to exposed H bond donors and acceptors of bases not involved in Watson-Crick H-bonding interactions between complementary bases in the ds DNA.
Figure: Triple helix
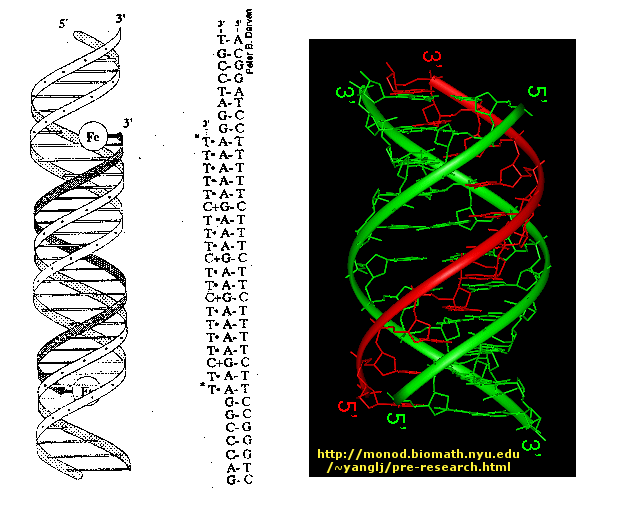
The single strand binds to such donors that are accessible in the major grove of dsDNA.
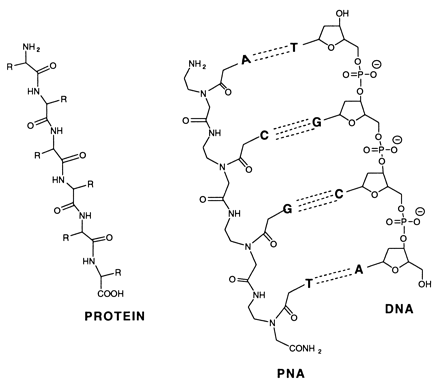
Peptide-nucleic acid analogs, as shown below, are promising new drugs. They are less charged than nucleic acids (so can more easily cross a membrane) and are resistance to cleavage by proteases and nucleases. One could image them binding to specific nucleotide sequences and inhibiting processes like DNA replication and transcription.
Transcription factors that are generally activated by genetic events or upstream signaling pathways are key regulators of cell state. Due to the extensive protein-protein interfaces and general absence of hydrophobic pockets that might inhibit protein:protein interactions required for transcriptional regulation, it has been difficult to design drugs that bind to transcription factors and modulate their activity. Moellering et al. report a successful development of a direct-acting antagonist of an oncogenic transcription factor, NOTCH1. This antagonist consists of cell-permeable stabilized α-helical peptides, SAHMs that was "stapled" into a stable helix through addition of two unnatural alkenyl amino acids which through ring closure sterically restrained the peptide in an alpha helix. The helix mimicked one protein:protein interface region in the ternary complex of DNA:NOTCH1:MAML1 (which is a coactivator protein). The peptides antagonized on NOTCH signaling and cell proliferation in T-cell acute lymphoblastic leukemia cells (T-ALL).
E6. DNA Binding and Genomic Analyses
Huge numbers (100,000 to 1 million) of different DNA molecules can be covalently attached to silicon or glass chips, as described above for the peptides. These sequences are located at specific x,y coordinates on the chip. DNA probes can then be made from cells (by adding reverse transcriptase to isolated mRNA. forming cDNA), which are then labeled with a fluorescent molecule. Easier yet, mRNA can be isolated from the cells and labeled with a fluorophore. When added to the chip, they will bind through complementary H-bond interactions to specific complementary DNA on the chip. Using this technique, an individual's entire genome could be analyzed in a short amount of time. For example, mutations in certain genes associated with cancer might be detected by fluorescent-DNA probes made from a possible mutant gene to specific DNA molecules in the chip that were designed to bind to mutant probes.
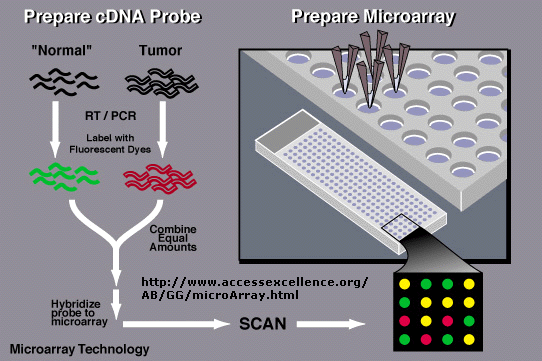
In an amazing variation, mRNA can be extracted from two different cells, a control and a tumor cell. The control mRNA can be labeled with a green fluorophore, while the tumor cell mRNA can be labeled with a red fluorphore. They can both be added to the chip containing a library of human genes. If the gene is expressed in both cell types, both types of labeled mRNA will bind and the spot on the chip will appear yellow.
If the gene is not expressed in either tissue, the spot will appear black. Genes that are only expressed in tumor cells will appear red and in control cells green. In a single experiment, the differential expression of genes in tumor cells can be determined. In this way, tumor-specific proteins can be identified, which could lead to the development of a vaccine against those tumor antigens. A typical microarray analysis for this type of experiment is shown below. (from Nature, 403, 699 (2000).
Figure:
![]() Detection
of Differential Gene Expression in Tumors and Normal Cells Using Microarray
Chip
Detection
of Differential Gene Expression in Tumors and Normal Cells Using Microarray
Chip
Array technologies have continued to evolve. Affymetrix has developed a array chip that contains over 38,000 genes, representing the entire human genome.
E7. RNA Binding Drugs
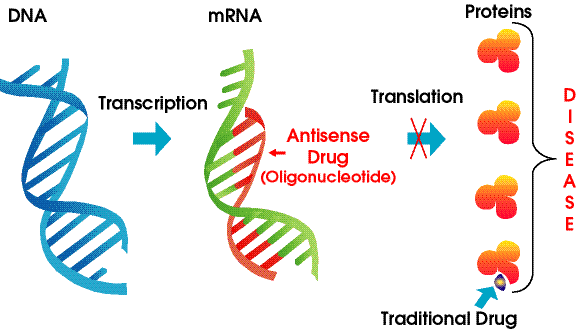
One of the best new alternatives is to design a RNA molecule complementary to a given mRNA for a specific protein selected for inhibition. This antisense RNA forms a dsRNA when it binds to the mRNA and inhibits translation of the mRNA.
Figure: antisense RNA forms a dsRNA
Cells can be altered through genetic engineering to make an antisense RNA within the cells by inserting an inverted copy of the target cDNA for the gene of interest into the cell, and allowing the cell to manufacture its own drug!
Figure: Cells can be altered through genetic engineering to make an antisense RNA within the cells
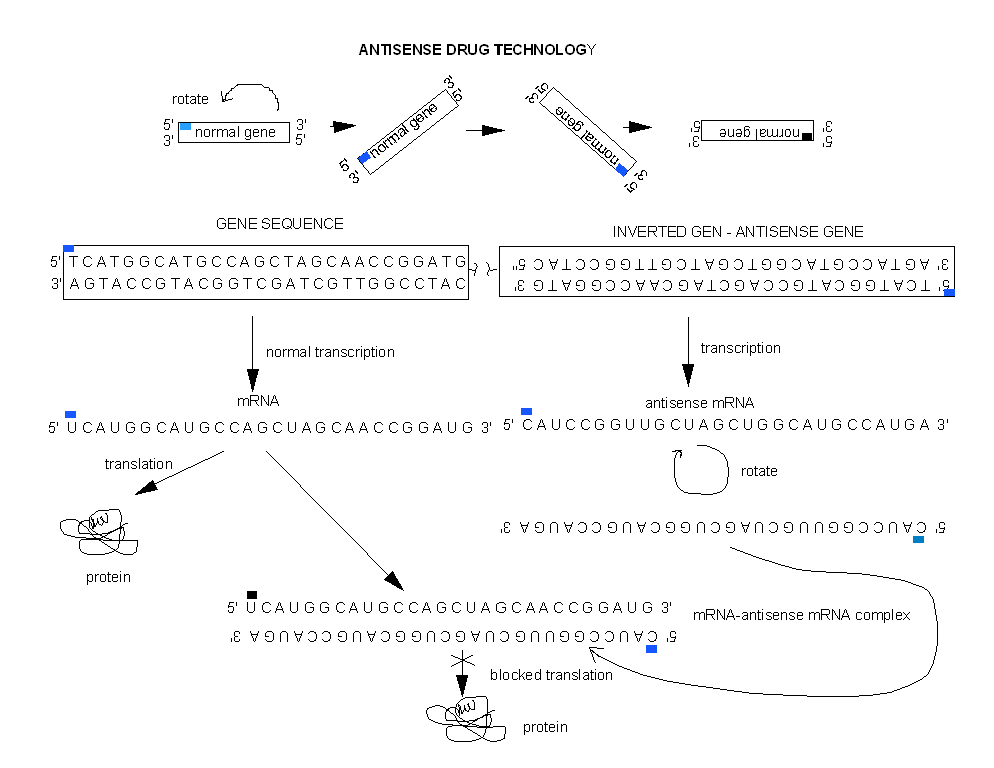
Even more exciting is the use of RNAis (RNA inference) for disease therapy. In contrast to traditional drugs, RNAis are one of nature's proven methods to inhibit gene transcription or RNA translation (mainly from viruses). Even though it was only discovered five years ago, it may be only a few years before drug trials based on RNAi take place. Especially promising are trials for siRNAs that inhibit HIV. To show that it works in organisms, mice were infected with a fusion of hepatitis C gene segment and a gene for a fluorescent protein luciferase. Specific siRNAs-treated mice showed a dramatic decrease of fluorescence, but not those treated with a nonspecific RNAi. As with other drugs, getting them into target cells is proving difficult. Simple liposome-encapsulated siRNAs apparently are not as effective as hoped. Carrying the siRNA into the cells with virus is yet another idea. Some companies pursuing this technology are shown below.
-
Antisense
drugs: AVI BioPharma
-
Calando:
RONDEL (RNAi/Oligonucleotide Nanoparticle Delivery) in which siRNA
is bound to a block polymer of a positive polymer interspersed among
cyclodexdrins, protecting the. siRNA from degrations by nucleases.
These self-associate to form nanopartaicles. Adamantine (hydophobic)
linked to polyethylene glycol (polar) to which targeting proteins (like
transferrin) are attached are added to the nanoparticles, leading to
PEG-dependent stabilization of the nanoparticle (and si-RNA) and
transferrin-dependent delivery to target cancer cells, which over
express transferrin receptors.
E8. Multivalent Inhibitors
In the previous study guide on binding and transcription, we discussed how a protein might bind DNA cooperatively if it interacts with a pre-bound protein adjacent to its normal binding site as well as its own binding site. The protein-protein interaction effectively reduces the apparent koff of the protein for its DNA binding site, which decreases the apparent Kd. Imagine a variation on this. Consider what might happen if a ligand for a macromolecule receptor found on the cell membrane was present in multiple copies. That is, what if the ligand was multivalent? What if the receptor was also multivalent? Both these cases are often found when complex ligands (bearing complex carbohydrates, for example) are present on the presenting ligand (a virus or bacteria, for example) and the macromolecule receptor is a cell surface protein. An interaction between the multivalent ligand and the receptor is necessary for the initial biological interaction between them.
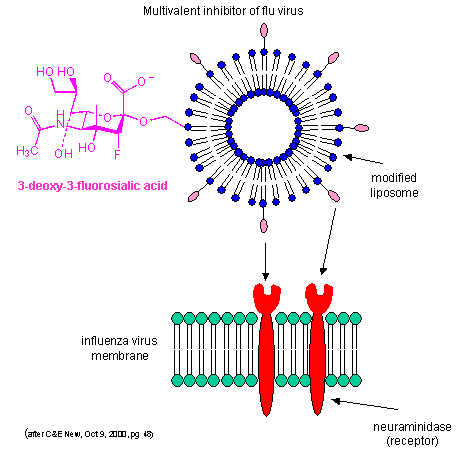
By decreasing the apparent koff as described above, multiple weak binding interactions can be effectively turned into strong interactions. Creating multivalent drug inhibitors of viral or bacterial interactions with cell membrane receptors would increase their efficacy. For example, consider the binding of the influenza virus to its receptor on a cell. Two viral proteins, hemagglutinin, as well as neuraminidase, binds to sialic acid residues on the glycoproteins receptors of the cells. A single, monovalent sialic acid derivative (acting as a drug) would be a low affinity inhibitor of the viral-cell interaction. A multivalent inhibitor, in which multiple sialic acid derivatives were covalently attached to a liposome proved to be a much more effective inhibitor since it could bind to multiple hemagglutinin and neuramindase molecules on the viral surface with an apparent lower Kd. This inhibitor is 1000X as potent as the monovalent inhibitor. This technique can be used not only to inhibit, but also activate biological reactions.
Figure: Multivalent inhibitor of flu virus
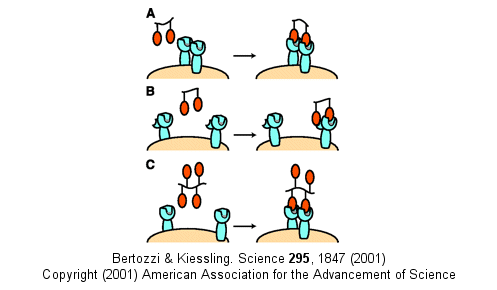
Other mechanism can also account for the amplified effects of multivalent ligands in addition to the effects on koff (which is related to the effective local concentration of the ligand at the receptor site - an effect also called the chelate effect). These include ligand-induced clustering of receptors on the cell surface and binding of multivalent ligands to more than one site or subsite on the receptor. These are illustrated below.
Figure: Multivalent Ligand Binding Models
E9. Targeted Delivery of Drugs to Tumors
The toxic effects of chemotherapeutic drugs might be eliminated if the drugs could be targeted to tumor cells and not to normal cells. Tumor cells presumably display unique protein molecules (tumor antigens) on their cell surface membranes that are not found on normal cell membranes. Drugs must enter the circulatory system of the specific targeted organ before they can be taken up by that organ. The endothelial cells that line the inner surface of blood vessels must display different proteins depending on the organ they are in, since circulating cells that interact with blood vessels in a specific organ appear to bind only to the endothelial cells in the specific organ. These cell and tissue-specific endothelial cell surface receptors impart specificity in binding. A recent study by Oh et al. demonstrated the presence of different proteins on endothelial cells found in blood vessels of normal mouse lungs and lung tumors. They injected silica particles into the blood. The particles bound to endothelial cells membranes. They homogenized, centrifuged and purified membrane-fractions bound to the silicon particles. The membranes were highly enriched in endothelial cells and caveolae, which are small indentations into the membrane and represent sites for the uptake of molecules, viruses, etc. into the cell. Molecular targets (such as viruses) could bind to receptors in the caveolae, and then through a process of invagination be taken up into the cell. The inner leaflet of the caveolae is lined with a protein called caveolin. Two endothelial cell transmembrane proteins, aminopeptidase P and annexin A1 were found specifically in normal and tumor lung endothelial cells, respectively. When given to rats with lung tumors, radioactive-labeled antibodies to annexin A1prolonged the life span of the rats.
E10. Diseases of Protein Misfolding and Aggregation
As we discussed earlier, many diseases are caused by protein misfolding, which is often associated with aggregation. (See What's New in Protein Chemistry: Protein Aggregates - Not Just Junk.) Neurological diseases associated with these processes include Jacob-Kreutzfeld disease (associated with prion proteins) and Alzheimer's and other amyloid diseases such as Familial amyloidotic polyneuropathy (FAP). In FAP, transthyrein, which normally exists in blood as a homotetramer dissociates into monomers which can aggregate into fibrils. Val30Met and Leu55Pro mutations promote dissociation of the tetramer and formation of aggregates. Conversely, Thr119Met inhibits tetramer dissociation. The aggregates deposit in heart, lungs, kidney, etc, leading to death. Hammaarstrom et al. have shown that the Thr119Met mutation increases the free energy of the transition state for the tetramer to monomer equilibrium, inhibiting that reaction and subsequent aggregation of the monomers. Amino acid 119 is at the dimer interface, consistent with these findings. They also have developed small molecule inhibitors of aggregation which appear to bind preferentially to the tetrameric state, increasing the activation energy for the transition state to the monomer.
There are many possible ways to prevent protein aggregation that work at various stages along the pathway of aggregation. For instance, in Alzheimers disease, secretase inhibitors would decrease the concentrations of amyloid beta (Aβ) protein, inhibiting its aggregation. Another possibility is to develop ligands (either small or large) that would bind either to the variant form of the protein (a partially unfolded or molten globule form of the protein) which would shift the equilibrium toward the variant monomer form, or to the normal monomer form of the protein, which would shift the equilbrium away from the variant form that aggregates. An example of this has been been described. Dumoulin et al. develop an camelid (camels and llamas) antibody against the normal human lysozyme. (Camelid antibodies were chosen since they are smaller than other vertebrate antibodies and may have a better chance of actually getting into cells and binding target proteins.) These antibodies, when added to a mutant form of lysozyme (Asp67His) which aggregates and leads to systemic lysozyme amyloidosis, prevented aggregate formation. Presumably the antibody to the normal conformation of lysozyme shifted the equilibrium of the mutant away from the misfolded, molten-globule like variant, to the normal conformation. The antibody binds to two regions of secondary structure (C-terminal region of the β-domain and the C-helix of the α-domain of the protein), not to the section of the protein that was mutated. In addition, the antibody interacted with only 11 of the 60 residues of the destablized region. X-ray crystal structures of the antibody-normal protein complex showed that the conformation of the normal protein did not change on antibody binding. The data supports the ideas that the antibody did not simply mask the destabilized structure and prevents its unfolding, but rather it promoted normal cooperative interactions within the whole protein necessary for normal protein folding.
Another type of neurodegenerative diseases is associated with aggregation of proteins that have aberrant stretches of Gln residues, caused by an expansion of the CAG coding sequence in genes for these "polyglutamine-containing" proteins. An example of such as disease is Huntington's Disease. The aggregates contain monomers with extensive beta-structure characteristic of amyloid disease. The small inhibitor dye, Congo Red, can bind to these beta structures and in vitro can dissociate existing aggregates and prevent aggregate formation by monomers. Sanchez et al. have recently shown that Congo Red has the same effect in mice models of Huntington's disease, and led to significant improvement in mice with preexisting symptoms.
E11. Links and References
![]() DrugBank
from the University of Alberta
DrugBank
from the University of Alberta
![]() DrugBank:
database is a unique bioinformatics and cheminformatics resource that
combines detailed drug (i.e. chemical, pharmacological and pharmaceutical)
data with comprehensive drug target (i.e. sequence, structure, and pathway)
information
DrugBank:
database is a unique bioinformatics and cheminformatics resource that
combines detailed drug (i.e. chemical, pharmacological and pharmaceutical)
data with comprehensive drug target (i.e. sequence, structure, and pathway)
information
![]() MOAD:
Mother of All Databases - largest collection of well resolved protein
crystal structures with clearly identified biologically relevant ligands
annotated with experimentally determined binding data
MOAD:
Mother of All Databases - largest collection of well resolved protein
crystal structures with clearly identified biologically relevant ligands
annotated with experimentally determined binding data
extracted from
literature
References
- Moellering, R. et al. Direct inhibition of the NOTCH transcription factor complex. Nature 462, 171 and 182 (2009)
- Keiser, M. et al. Predicting new molecular targets for known drugs. Nature 462, 167 and 175 (2009)
- Oh, P. et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 429, pg 629 (2004)
- Dumoulin, M. et al. A camelid antibody fragment inhibits the formation of amyloid fibrils by human lysozyme. Nature, 424, pg 783 (2003).
- Huang, D. et al. Crystal structure of NF-κB (p50)2 complexed to a high affinity RNA aptamer. Proc. Natl. Acad. Sci. USA. 100, pg 9268 (2003)
- Sanchez, I. et al. Pivotal Role of oligomerization in expanced polyglutamine neurodegenerative disorders. Nature. 421, pg 373, (2003)
- Hammarstrom, P. et al. Prevention of Transthyretin Amyloid Disease by changing protein misfolding energetics. Science. 299, pg 713 (2003)
- Rusconi, C. et al. RNA apatmaers as reversible antagonists of coagulation factor IXA. Nature. 419, pg 90, 23 (2002)
- Zasloff. Antimicorbial peptides of multicellular organisms. Nature. 415, pg 389 (2002)
- RNA Interference: The Short Answer (blocking mRNA). Nature. 411 pg 428 (2001)
- Fernandez-Lopez et al. Rings of Destruction. (Cyclic peptides as drugs). Natuare. 412 pg 392, 452 (2001)
- Small but mighty timekeepers. (small RNA inhibition of translation) Nautre. 408, pg 37 (2000)
- Shokat et al. A Chemistry Set for Life. (Chemical genetics: inhibitors designed for specific gene products)*** Nature 407, pg 282, 395 (2000)
- Clark et al. Molecular Switches in Metastasis. Nature. 406. pg 466, 532 (2000)
- Root et al. Protein Design of an HIV-1 Entry Inhibitor. Science. 291, pg 884 (2001)
- Tyler et al. Peptide nucleic acids targeted to the neurotensin receptor and administered i.p. cross the blood-brain barrier and specifically reduce gene expression . PNAS, Vol. 96, Issue 12, 7053-7058, June 8, 1999.
- Interfering with Gene Expression (silencing at the mRNA level). Science. 288, pg 1370 (2000)
- Gellman et al. Slaying Bacteria with β-peptides. Nature. 404, pg 565 (2000)
- RNA interference (RNAi by short RNAs that demonstrate transcriptional silencing). Science. 287, pg 2431, 2494 (2000)
- Evans and Relling. Pharmocogenomics: Translating function genomics into rational therapeutics. Science. 286, pg 487 (1999)
- Kiessling, L et al. Synthetic multivalent ligands in the exploration of cell-surface Current Opinion in Chemical Biology, 4, pg696 (2000)
 Navigation
Navigation
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 5E: Modern Methods in Drug Development

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.