Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 5 - BINDING
A: INTRODUCTION TO REVERSIBLE BINDING
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Update: 3/25/16
|
Learning Goals/Objectives for Chapter 5A: After class and this reading, students will be able to
|
A7. Molecular Basis of High Affinity Interactions
What differentiates high and low affinity binding at the molecular level? Do high affinity interactions have lots of intramolecular H-bonds, salt bridges, van der Waals interactions, or are hydrophobic interactions most important? Recently, the crystal structures of a variety of antibody-protein complexes were determined in order to study the basis of affinity maturation of antibody molecules. It is well know that antibodies elicited on exposure to a foreign molecule (antigen) are initially of lower affinity than antibodies released later in the immune response. An incredible number of different antibodies can be made by antibody-producing B cells due to genetic mechanisms (combining different variable regions of antibody genes through splicing, imprecise splicing, and hypermutation of critical nucleotides in the genes of antigen binding regions of antibodies). Clones of antibody-producing cells with higher affinity are selected through binding and clonal expansion of these cells. Investigators studied the crystal structure of 4 different antibodies which bound to the same site (epitope) on the protein antigen lysozyme. Increased affinity was correlated with increased buried apolar surface area and not with increased numbers of H bonds or salt bridges.
Table: Characteristics of Antibody:Hen Egg Lysozyme Complexes(HEL)
| Antibody | H26-HEL | H63-HEL | H10-HEL | H8-HEL |
| Kd (nM) | 7.14 | 3.60 | 0.313 | 0.200 |
| Intermolecular Interactions | ||||
| H bonds | 24 | 25 | 20 | 23 |
| VDW contacts | 159 | 144 | 134 | 153 |
| salt bridges | 1 | 1 | 1 | 1 |
|
Buried Surface Area |
||||
| ΔASURF (A2) | 1,812 | 1,825 | 1,824 | 1,872 |
| ΔASURF-polar (A2) | 1,149 | 1,101 | 1,075 | 1,052 |
| ΔASURF-apolar (A2) | 663 | 724 | 749 | 820 |
Li,Y. et al. Nature: Structural Biology. 6, pg 484 (2003)
Electrostatic interactions between biological molecules are still very important interactions, even though we may consider them to be nonspecific. Witness the interaction of DNA binding proteins with positive domains with the negative polyanion, DNA. The initial encounter will be electrostatic in origin and obviously important to targeting the proteins to DNA where additional specific interactions may take place.
In a similar example (Yeung, T et al.), it was recently reported that moderately positively charged proteins are directed to endosomes and lysosomes through interactions with negatively charged membrane phosphatidylserine (PS), whereas more positively charged proteins are targeted to the inner surface of the plasma membrane, which is enriched in PS and phosphorylated phosphatidyl inositol derivatives (PIP2, PIP3), as shown below.
Figure: negatively charged phospholipids in biological membranes
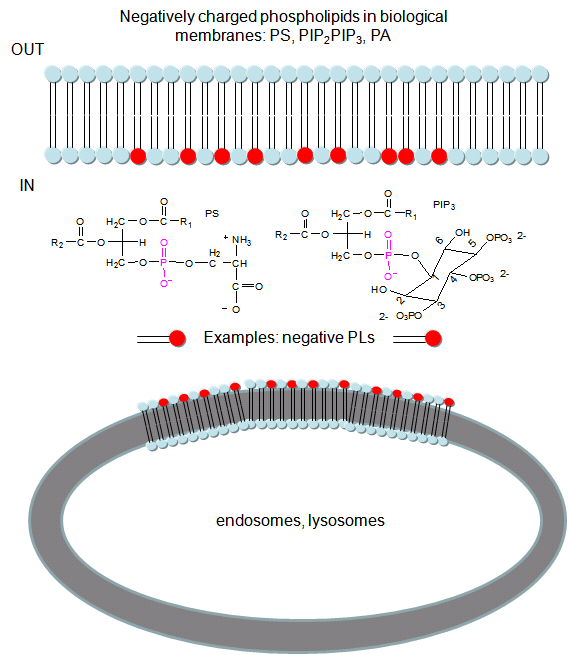
To study this they used the C2 domain of lactadherin (Lact-C2) from milk that binds PS in the presence of calcium. The C2 domain was covalently linked to the green fluorescent protein, a protein which contains an internal fluorophore comprised of three amino acids (Ser65–Tyr66–Gly67) that cyclize spontaneously on folding to produce a fluorophore which emits green light. A fusion gene of Lact-C2 and GFP was introduced in wild type (WT) and mutant yeast lacking PS. It was bound to the inner leaflet in WT cells and to endosome and lysosome vesicles , but found diffused through cytoplasm in mutant cells. They also made cationic probes with farnesyl tails attached which could anchor the soluble probes to membranes. The most positively charged probes were recruited to the plasma membrane inner leaflet, while less charged ones were recruited to internal vesicles. The authors speculate that PS on cytoplasmic membrane layers can target signal transduction proteins to these regions.
 Antibodies with Infinite Affinity. Chmura et al. PNAS. 98, pg 8480
(1998)
Antibodies with Infinite Affinity. Chmura et al. PNAS. 98, pg 8480
(1998)
Docking
The quantitative methods described above do not elucidate the mechanism of binding. Computer programs have been developed that allow the docking of a ligand (small molecule or even another protein) to another protein. The automatic docking of flexible ligands to proteins can be modeled using Autodock. AutoDock contains the following programs:
-
AutoDock performs the docking of the ligand to a set of grids describing the target protein;
-
AutoGrid pre-calculates these grids;
-
AutoTors sets up which bonds will treated as rotatable in the ligand.
Molecular dynamics simulations can also be used to study the actual binding and unbinding processes.
The Crowded Cell
Most binding studies are performed in vitro with dilution concentrations of both macromolecule and ligand. Are these conditions illustrious of conditions inside a cell? The answer is no! Cells are very crowded with organelles, macromolecular complexes, and cytoskeletal components which provide internal architecture to the cell, etc. Total macromolecule concentration in the cell has been estimated to be as high as 400 g/1L = 400 g/1000 mL = 0.4 g/mL = 400 mg/mL. Try to dissolve a water soluble protein like albumin to those concentrations! From 5 to 40% of the entire cellular volume is occupied with large molecules, and at the upper range, very little space exists for other large macromolecules.
 Navigation
Navigation
Return to Chapter 5A: Introduction to Reversible Binding Sections
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 5A: Introduction to Reversible Binding

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.