Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 5 - BINDING
E: MODERN METHODS IN DRUG DEVELOPMENT
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Updated: 03/31/16
|
Learning Goals/Objectives for Chapter 5E: After class and this reading, students will be able to
|
E2. Combinatorial Drug Development
Instead of searching various organisms to find a possible drug from all the extractable molecules of the organism, people are now synthesizing an enormous number of randomly constructed molecules, and then selecting active molecules from this random pool by binding or other bioassays. Solid phase synthesis of random peptides or nucleic acids can be used to generate thousands of possible test inhibitors. Those peptides or oligonucleotides that bind flurorescent-labeled protein can be determined, as shown below.
Figure: Combinatorial Drug Development.
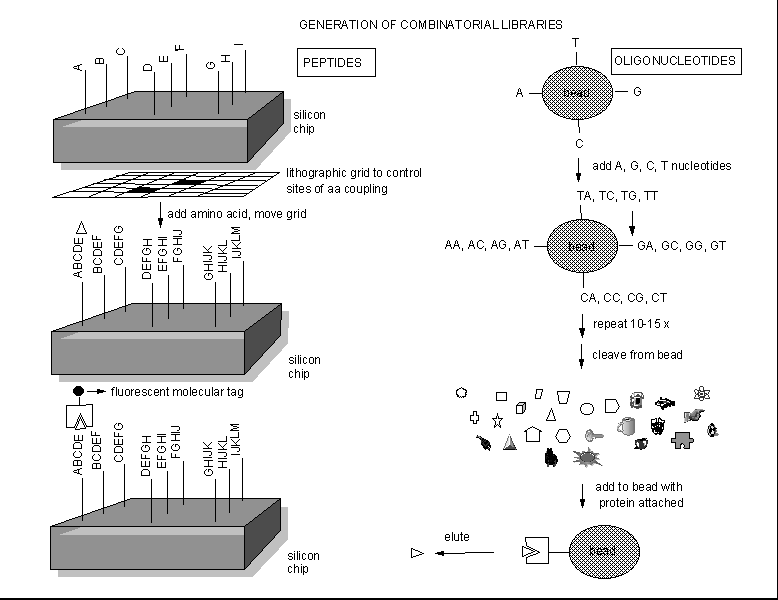
These structures can then be decorated or used to design peptide or oligonucleotide mimetics, which would be more resistant to enzymatic degradation. Another way to make a large library of peptide inhibitors is to make them using genetic engineering.
Recently, peptides made of D-amino acids have been synthesized. These are much more resistant to proteases than normal L-amino acid peptides. D amino acid peptides can only be made in the lab, and not through genetic engineering. An intriguing variation of this technique has recently been developed. Investigators have synthesized entire proteins in the lab using D-amino acids. Combinatorial L-peptide libraries were made using genetic engineering in bacteria. L-peptides were selected that bound with high affinity to the D-protein. The corresponding D-peptides were made in the lab, which then were found to bind to the normal L-protein.
In another combinatorial technique, single stranded RNA molecules called aptamers can be synthesized and tested for the binding/inhibitory activity for a enzyme. RNA molecules, which can form complex secondary and tertiary structure, can also present, given the appropriate sequence, a complementary binding surface to sites on proteins. Aptamers with high affinity are sequenced. Bases that appear to be necessary for high affinity binding are identified by comparing the sequence of different aptamers. This knowledge is then used to synthesize even tighter binding aptamers, in an process that mimics evolutionary selection for high affinity binding. Such a high affinity aptamer was recently made to bind to and inhibit Factor IXa, an active enzyme required for initiation and propagation of blood clotting. Traditional anticoagulant drugs are extremely valuable in treating and preventing inappropriate clot formation, which can lead to strokes (brain attacks) and heart attacks. The problem with these drugs is that they must not tip the clotting/anticlotting balance to far in the direction of clot prevention, since there are many times when clot formation is the appropriate biological response (such as in the prevention of hemorrhagic stoke). Rusconi et al. realized that once they had developed the high affinity aptamer for factor IXa binding, they immediately could synthesize an anedote for the aptamer. It would have a complementary sequence to the Factor IXa binding bases of the drug aptamer, that would allow it to bind to the original apatmer and form a double-stranded RNA sequence. The crystal structure of the homodimer transcription factor, NF-κB (p50)2 bound to an RNA aptamer has been determined. The RNA sequence is dissimilar to the DNA sequence which binds the transcription factor, even though both have similar dissociation constants.
![]() Jmol:
Updated Nf-Kb(P50)2
Complexed To A High- Affinity RNA Aptamer
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Nf-Kb(P50)2
Complexed To A High- Affinity RNA Aptamer
Jmol14 (Java) |
JSMol (HTML5)
 Navigation
Navigation
Return to 5E. Modern Methods in Drug Development Sections
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 5E: Modern Methods in Drug Development

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.