Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 7 - CATALYSIS
B: MECHANISMS OF ENZYME CATALYSIS
BIOCHEMISTRY - DR. JAKUBOWSKI
004/12/16
|
Learning Goals/Objectives for Chapter 7B: After class and this reading, students will be able to
|
B3. Chymotrypsin
Chymotrypsin, a protease, cleaves amides as well as small ester substrates after aromatic residues. The following data using different chymotrypsin substrates suggests a covalent intermediate occurs on chymotrypsin catalyzed cleavage of esters and amides.
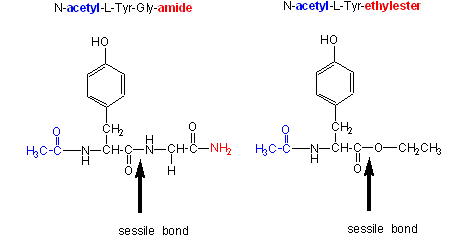
- changing the substrate - for example changing the leaving group or acyl sustituents of a hydrolyzable substrate:
| Chymotrypsin substrate cleavage, 25oC, pH 7.9 | |||
| kinetic constants | Acetyl-Tyr-Gly-amide | Acetyl-Tyr-O Ethylester | Ester/Amide |
| kcat (s-1) | 0.50 | 193 | 390 |
| Km (M) | 0.023 | 0.0007 | 0.03 |
| kcat/Km (M-1s-1) | 22 | 280,000 | 12,700 |
|
Kinetic constants for chymotrypsin cleavage of N-acetyl-L-Trp Derivatives - N-acetyl-L-Trp-X |
||
| X | kcat (s-1) | Km x 103 (M) |
| -OCH2CH3 | 27 | 0.097 |
| -OCH3 | 28 | 0.095 |
| -p-nitrophenol | 31 | 0.002 |
| -NH2 | 0.026 | 7.3 |
- the kcat and kcat/Km are larger and the Km smaller for ester substrates compared to amide substrates, suggesting that amides are more difficult to hydrolyze (tables 1 and 2 above). This is expected given the poorer leaving group of the amide.
- the kcat for the hydrolysis of ester substrates doesn't depend on the nature of the leaving group (i.e. whether it is a poorer leaving group such as methoxy or a better leaving group such as p-nitrophenolate) suggesting that this step is not the rate limiting step for ester cleavage. (Table 2). Without the enzyme, p-nitrophenyl esters are cleaved much more rapidly than methyl esters. Therefore deacylation must be rate limiting. But deacylation of what? If water was the nucleophile, release of the leaving group would result in both products, the free carboxyl group and the amine being formed simultaneously. This suggests an acyl-enzyme covalent intermediate.
- When the acyl end of the ester substrate is changed, without changing the leaving group (a p-nitrophenyl group), a covalent intermediate can be trapped. Specifically, the deacylation of a trimethyacetyl group is much slower than an acetyl group. It is so slow that a 14C-labeled trimethylacetyl-labeled chymotrypsin intermediate can be isolated after incubation of chymotrypsin with 14C-labeled p-nitrophenyltrimethylacetate using gel filtration chromatography.
We have seen a kinetic mechanism consistent with these ideas before. The reaction equations are shown below:
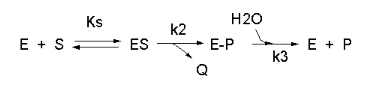
In this reaction, a substrate S might interact with E to form a complex, which then is cleaved to products P and Q. Q is released from the enzyme, but P might stay covalently attached, until it is expelled. This conforms exactly to the mechanism described above. For chymotrypsin-catalyzed cleavage, the step characterized by k2 is the acylation step (with release of the leaving group such as p-nitrophenol in Lab 5). The step characterized by k3 is the deacylation step in which water attacks the acyl enzyme to release product P (free phosphate in Lab 5). In class and for homework you derived the following equation::
Equation 7: v = [(k2k3)/(k2 + k3)]EoS/[Ks(k3)/(k2+k3)] + S
As mentioned above, for hydrolysis of ester substrates, which have better leaving groups compared to amides, deacylation is rate limiting, ( k3<<k2). Then equation 7 becomes
v = k3EoS/[Ks(k3)/(k2) + S]
Vm = k3Eo and Km = Ks(k3)/(k2)
For amide hydrolysis, as mentioned above, acylation can be rate-limiting (k2<<k3). Then equation 7 becomes:
v = k2EoS/[Ks+ S]
Vm = k2Eo and Km = Ks
Just as we saw before for the rapid equilibrium assumption (when ES falls apart to E + S more quickly than it goes to product), Km = Ks in amide hydrolysis.
- changing the pH or ionic strength - which can give data about general acids/bases in the active site:
- a graph of kcat as a function of pH indicates that a group of pKa of approx. 6 must be deprotonated to express activity (i.e. Vm/2 is at about pH 6). This suggests that an active site histidine is necessary, which if it must be deprotonated to express activity, must be acting as a general base.
- a graph of kcat/Km shows a bell-shaped curve indicating the necessity of a deprotonated side chain with a pKa of about 6 (i.e. the same His above) and a group which must be protonated with a pKa of about 10. This turns out to be an N terminal Ile (actually at the 16 position in the inactive precursor of chymotrypsin called chymotrypsinogen, which on activation of chymotrypsinogen loses the first 15 amino acids by selective proteolysis), which must be protonated to form a stabilizing salt bridge in the protein.
Note: A new theoretical computer program,
called
THEMATICS (theoretical microscopic titration curves) has been developed
to calculate the titration curves for all ionizable groups in a protein.
When performed on test proteins, those amino acids that showed anomalous
curves (flattened compared to normal titration curves) where usually found
in the active site of the protein. The flattened curves show that the
amino acid is partially protonated over a wider range of pH then
theoretically expected. The program can be used to predict active site
regions on protein of known structure but unknown function, and will be
useful in the emerging field of proteomics. (figure below from Proc. Natl.
Acad. Sci. USA, Vol. 98, Issue 22, 12473-12478, October 23, 2001)
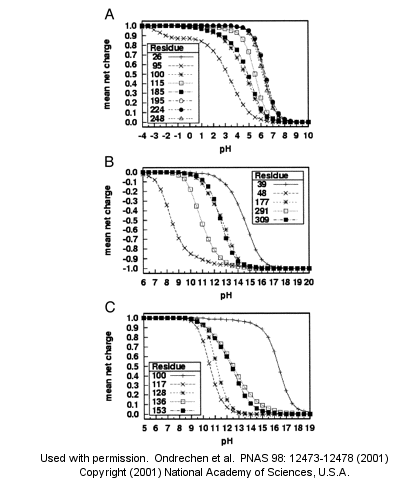
Fig. 1. Sample theoretical titration curves. Predicted mean net charge as a function of pH. (A) All of the histidine residues in the A chain of TIM: His-26 (+), His-95 (�), His-100 (*), His-115 (open square), His-185 (filled square), His-195 (open circle), His-224 (filled circle), and His-248 (). (B) Selected tyrosine residues of AR: Tyr-39 (+), Tyr-48 (�), Tyr-177 (*), Tyr-291 (open square), and Tyr-309 (filled square). (C) Selected lysine residues of PMI: Lys-100 (+), Lys-117 (�), Lys-128 (*), Lys-136 (open square), and Lys-153 (filled square). TIM, triosephosphate isomerase; AR, aldose reductase; PMI, phosphomannose isomerase. Remember that depending on the protein microenvironment, the pKa of a side chains like Asp can vary from 0.5 to 9.2!
- the enzyme - by chemical modification of specific amino acids, or through site-specific mutagenesis:
- modification of chyrmotrypsin (and many other proteases) with diisopropylphosphofluoridate (DIPF) modifies one of many Ser residues (Ser 195), suggesting that it is hypernucleophilic. and probably the amino acid which attacks the carbonyl C in the substrate, forming the acyl-intermediate.
Figure: diisopropylphosphofluoridate
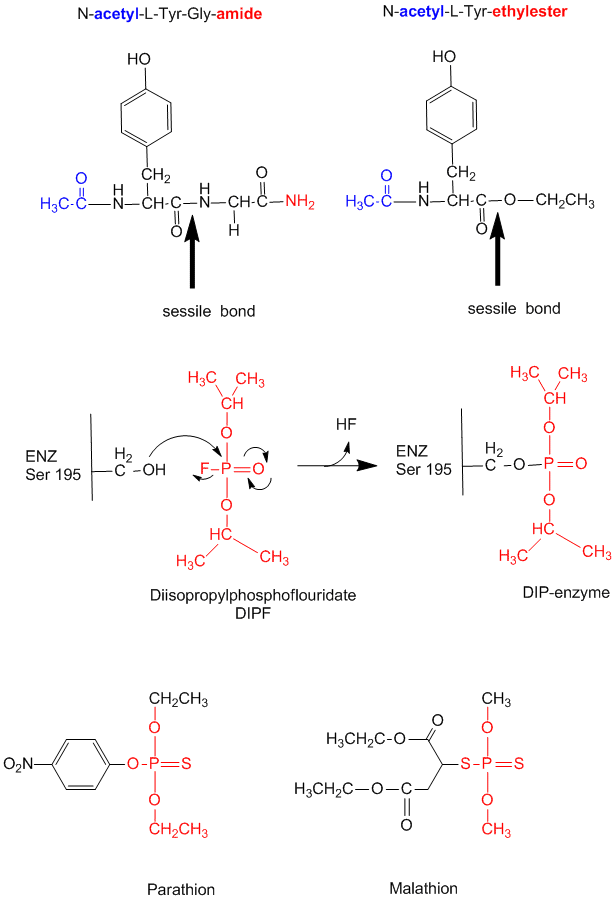
Malathion and ethyl parathion (organic phosphate pesticides) have similar structures and reactivities as DIPF, and selectively inhibit serine proteases in insects.
- modification of the enzyme with tos-L-Phe-chloromethyl ketone inactives the enzyme with a 1:1 stoichiometry which results in a modified His.
Figure: tos-L-Phe-chloromethyl ketone
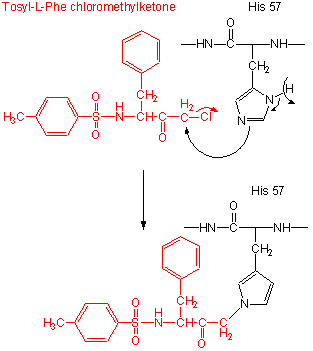
- comparison of the primary sequence of many proteases show that three residues are invariant: Ser 195, His 57, Asp 102.
- site-specific mutagenesis show that if Ser 195 is changed to Ala 195, the enzymatic activity is almost reduced to background.
Figure: Serine Protease Mechanism
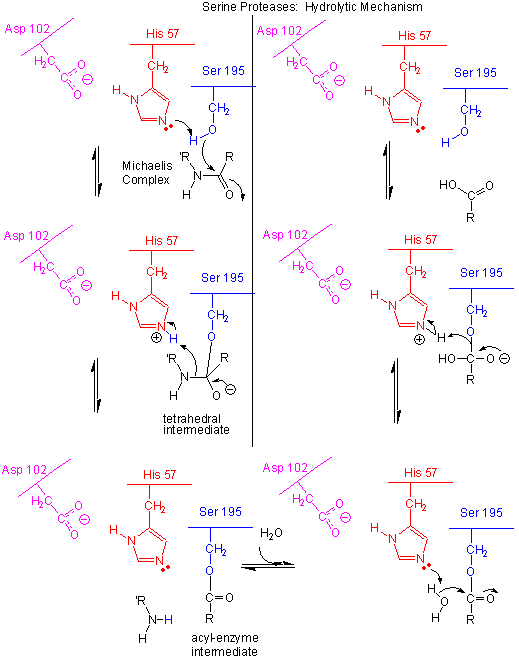
- The deprotonated His 57 acts as a general base to abstract a proton from Ser 195, enhancing its nucleophilicty as it attacks the electrophilic C of the amide or ester link, creating the oxyanion tetrahedral intermediate. Asp 102 acts electrostatically to stabilize the positive charge on the His.
- The oxyanion collapses back to form a double bond between the O and the original carbonyl C, with the amine product as the leaving group. The protonated His 57 acts as a general acid donating a proton to the amine leaving group, regenerating the unprotonated His 57.
- The mechanism repeats itself only now with water as the nucleophile, which attacks the acyl-enzyme intermediate, to form the tetrahedral intermediate.
- The intermediate collapses again, releasing the E-SerO- as the leaving group which gets reprotonated by His 57, regenerating both His 57 and Ser 195 in the normal protonation state. The enzyme is now ready for another catalytic round of activity.
- The mechanism for the first nucleophilic attack (by Ser) is the same as for the second (by water). The reverse mechanism of condensation of two peptide would be the reverse of the above mechanism, and is an example of the principle of microscopic reversibility.
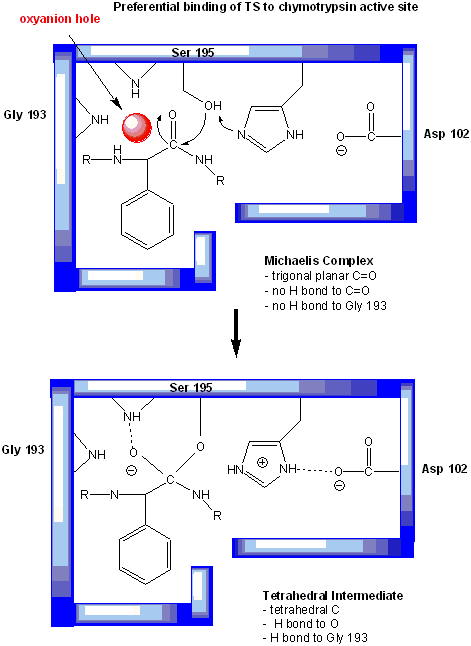
In short, all the catalytic mechanisms we encountered previously are at play in chymotrypsin catalysis. These include nucleophilic catalysis (with the Ser 195 forming a covalent intermediate with the substrates), general acid/base catalysis with His 57, and loosely, electrostatic catalysis with Asp 102 stabilizing not the transition state or intermediate, but the protonated form of His 57. An important point to note is that His, as a general acid and base catalyst, not only stabilizes developing charges in the transition state, but also provides a path for proton transfer, without which reactions would have difficulty in proceeding. One final mechanism is at work. The enzyme does indeed bind the transition state more tightly than the substrate. Crystal structures with poor "pseudo"-substrates that get trapped as partial tetrahedrally-distorted substrates of the enzyme and with inhibitors show that the oxyanion intermediate, and hence presumably the TS, can form H-bonds with the amide H (from the main chain) of Gly 193 and Ser 195. These can not be made to the trigonal, sp2 hybridized substrate. In the enzyme alone, the hole into which the oxyanion intermediate and TS would be placed is not occupied. This oxyanion hole is occupied in the tetrahedral intermediate.
Figure: oxyanion hole in serine proteases: TS stabilzation
Web Links:
![]() Jmol:
Updated Chymotrypsin:D-Leu-L-Phe-p-fluorobenzylamde complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Chymotrypsin:D-Leu-L-Phe-p-fluorobenzylamde complex
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol: Updated
Chymotrypin-Phenylethylboronate Inhibitor Complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
Chymotrypin-Phenylethylboronate Inhibitor Complex
Jmol14 (Java) |
JSMol (HTML5)
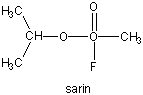
Many enzymes have active site serines which act as nucleophilic catalysts in nucleophilic substitution reactions (usually hydrolysis). One such enzyme is acetylcholine esterase which cleaves the neurotransmitter acetylcholine in the synapse of the neuromuscular junction. The transmitter leads to muscle contraction when it binds its receptor on the muscle cell surface. The transmitter must not reside too long in the synapse, otherwise muscle contraction will continue in an uncontrolled fashion. To prevent this, a hydrolytic enzyme, acetylcholine esterase, a serine esterase found in the synapse, cleaves the transmitter, at rates close to diffusion controlled. Diisopropylphosphofluoridate (DIPF) also inhibits this enzyme which effectively makes it a potent chemical warfare agent. An even more fluoride-based inhibitor of this enzyme, sarin, is the most potent lethal chemical agent of this class known. Only 1 mg is necessary to kill a human being.
Figure: sarin
 Navigation
Navigation
Return to 7B: Methods of Enzyme Catalysis
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 7B: Mechanisms of Enyzme Catalysis

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.