Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 7 - CATALYSIS
B: MECHANISMS OF ENZYME CATALYSIS
BIOCHEMISTRY - DR. JAKUBOWSKI
004/12/16
|
Learning Goals/Objectives for Chapter 7B: After class and this reading, students will be able to
|
B9. CRISPR-Cas 9
Introduction
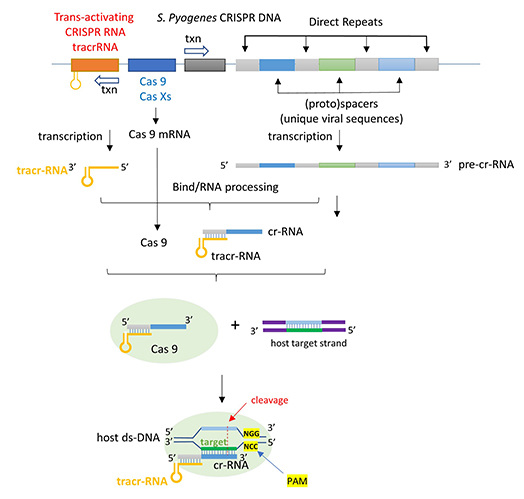
The CRISPR (clustered regularly interspaced short palindromic repeats) operon was initially discovered as part of the adaptive immune system of bacteria and archea, which must defend themselves against viruses (bacteriophages) and unwanted plasmid transferred from both bacteria. It would be ideal for bacteria to recognize previous exposure to viruses and their nucleic acids as the basis of their immunological memory system. Given the tendency of viral DNA to integrate into the host genome (which allows later transcription and translations of the viral genes in the process of new virus production), immunological memory could be based on that viral integrated DNA. Without going into detail, viral DNA can be integrated between two direct repeats in the bacterial genome. DNA from different viruses from previous exposures is also incorporated in the same fashion. One site of integration is the CRISPR operon. The DNA of the CRISPR operon contains both protein coding and noncoding regions which are transcribed and processed to form at least three RNA molecules (see figure below):
- a coding Cas 9 mRNA this is translated to produce the Cas 9 (CRISPR associated protein);
- a noncoding cr-RNA (CRISPR RNA)
- a noncoding tracr-RNA (trans-activating CRISPR RNA)
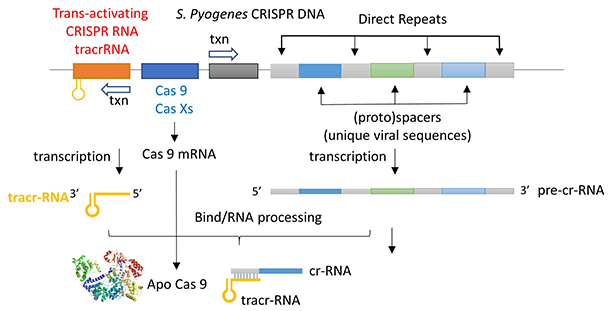
The two mature noncoding RNAs eventually associate to form a binary complex. When using CRISPR-Cas 9 in eukaroytic gene editing applications, the two noncoding RNAs are covalently combined into one large synthetic guide RNA (sg-RNA), described later in this section. The Cas 9 protein is an endonuclease that cleaves both strands of bound target dsDNA in a blunt-end fashion at specific sequences . This occurs after the DNA binds to two arginines (1333 and 1335) in Cas9 through a short (3-5+ bases) recognition protospacer adjacent motif (PAM) located three base pairs from the cleavage site. The DNA must also bind in a complementary and specific fashion to the protein-bound noncoding cr+tracr-RNAs (or a single sg-RNA molecule for gene editing applications). Binding and cleavage of target DNA would obviously render a recognized DNA from an invading bacteriophage inactive.
Basic research into the bacterial CRISPR system has led to revolutionary and explosive applications of this gene editing system in eukaroytes. The hope is that CRISPR technology will give us a precise and incredibly cheap way to do gene therapy in diseased cells and organisms.
We have discussed the structure and function of many proteins. Protein enzymes are key to life as they catalyze almost all biological reactions. Most key enzymes are regulated. The activity of Cas 9 must be carefully controlled. Think of the consequences if the enzyme were to cleave promiscuously at off-site targets! This section will help you understand several critical features of this enzyme:
- How does the enzyme find its correct target site, a 20 nucleotide DNA sequence and a proximal PAM site, among all the possible alternative sites. Think of how many PAM sequences there must be in the host DNA genome!
- How can the enzyme be "turned" on when it finds its target site and remain off when free, but more importantly, when it is bound off-site?
First we will discuss the apo- form of the enzyme without bound substrate and RNA.
ApoCas 9
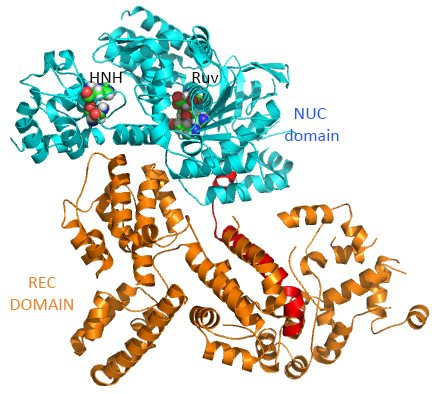
This section will focus on the Type II-A Cas9 from Streptococcus pyogenes (SpyCas9 or SpCas9). Cas 9 is an endonuclease that cleaves both strands of DNA 3 base pairs from a DNA motif, NCC/NGG, called PAM. It has two distinct lobes. The nuclease lobe (NUC), amino acids 1-56 and 718-1368, has two different nuclease domains for the two cleavages. The recognition or receptor lobe (REC), amino acids 94-717, interacts with the RNA molecules. There is also an arginine-rich bridge helix (57-93).
The enzyme has two catalytic nuclease domains:
- HNH-like nuclease domain which cleaves the "target" DNA strand, which is complementary to the RNA the confers specificity to the enzyme. The key catalytic residues are His 840 and Asn 854. It also contains a Mg ion;
- Ruv-like domain that cleaves the complementary "non-target" strand with key active site residues Asp 10, Glu 762, Asp 986 and His 983. It also contains a bound Mn ion. The two lobes are separated by two linkers, amino acids 712-717, and an arginine-rich bridge (basic helix - BH), amino acids 628-658.
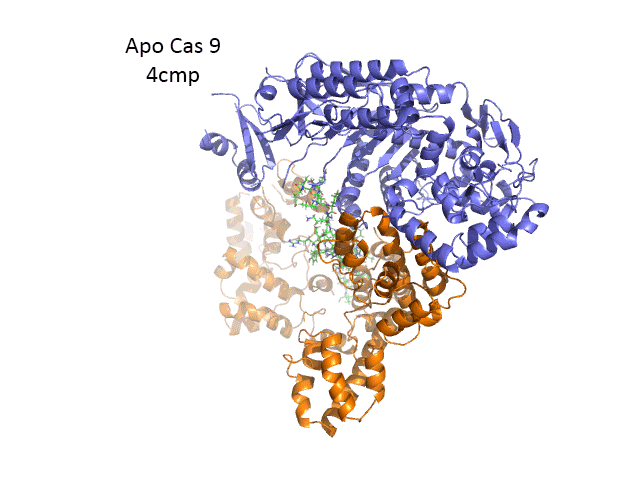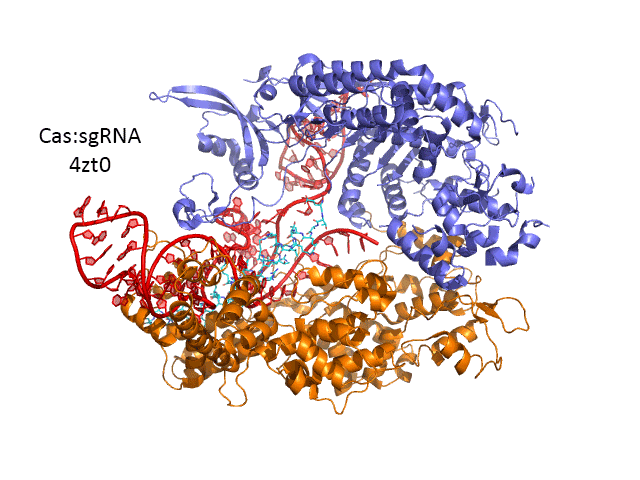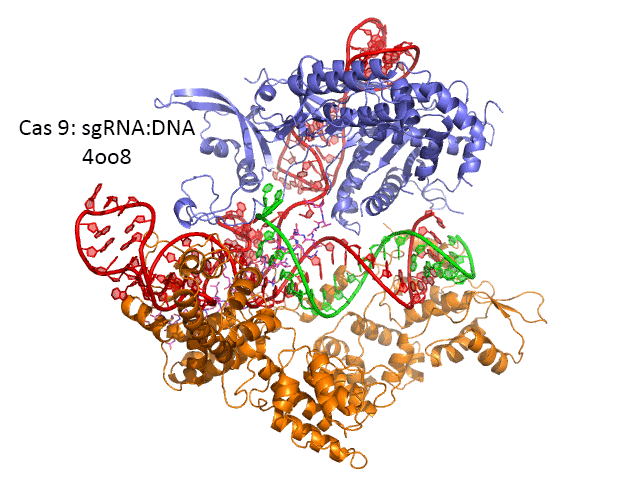
The overall structure of the apoenyzme (without bound RNA and DNA,pdb id 4cmp) is shown in the figure below, which shows the NUC domain (light blue) with the two catalytic domains (HNH and Ruv), the REC domain (orange) and the BH helix (red).
A close up view showing the two catalytic sites is shown in the figure and Jsmol below.
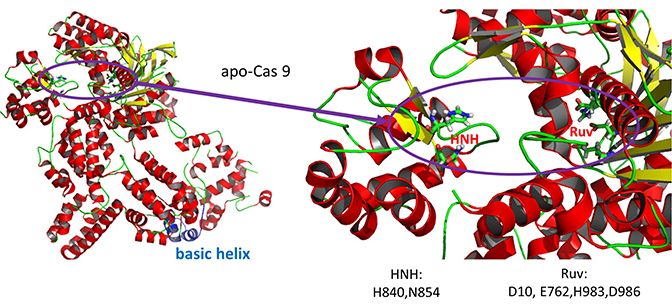
A Jsmol of both the apo and holo forms of the enzyme can be found at the link below.
![]() Jmol: Cas 9
(4cmp - apo and 4008 - holo)
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Cas 9
(4cmp - apo and 4008 - holo)
Jmol14 (Java) |
JSMol (HTML5)
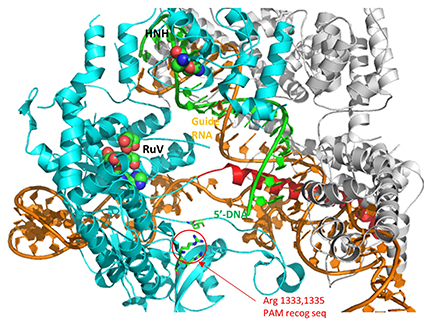
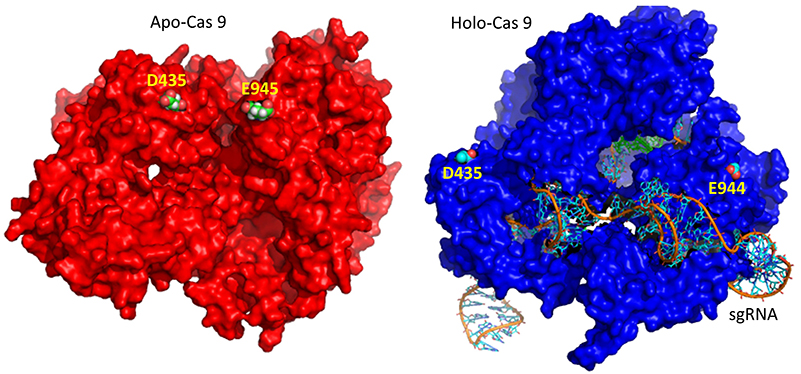
A comparison of the crystal structure of the apo-Cas 9 and the ternary Cas 9: sgRNA:DNA target strand complex shows a significant conformational change on binding nucleic acids. The structure of the holoenzyme (ternary complex) is shown in the figure and in the Jsmol below.
The extent of the conformation change between apo- and holo-Cas 9 enzymes can be seen by examining the distance between D435 and E 944/945 in the figure below. The importance of this change will be described later.
The figure below shows the pathway from transcription of the relevant CRISPR genes (coding and noncoding) to the assembly of the ternary complex and the blunt end cut of the target DNA strand three nucleotides from the PAM sequence.
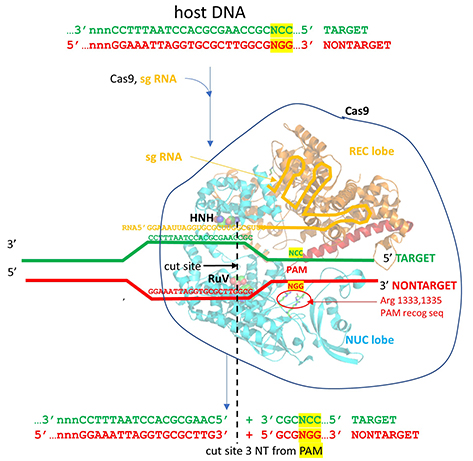
The image below shows an expanded view of the ternary complex.
Mechanism of DNA binding and cleavage
The above figures do not speak to the mechanism of the binding processes that form the ternary complex. Kinetic and structural studies have been conducted to elucidate the mechanism of binding and cleavage and address the following questions:
- which binds first, the RNA or DNA?
- What are the consequences of the profound conformational changes on formation of the ternary complex?
The specificity of target DNA binding depends both on enzyme:PAM DNA and enzyme:sgRNA (or tracr- and crRNA) interactions. It should seem improbable that the trinucleotide PAM DNA sequence (NGG in S. pyogenes), which interacts with a pair of arginines (R 1333, R 1335) through H-bonding, as shown in the images above, and other local sites in Cas 9 would provide the sole or even the majority of the binding interactions. The figure below shows the Args:PAM interaction (pdb code 4un3)
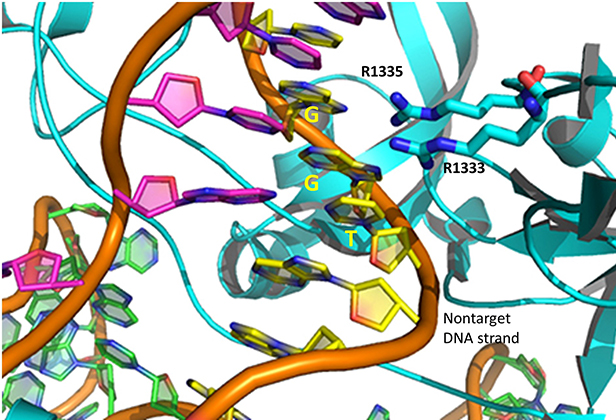
Hence it is most likely that RNA binds first. Indeed, it does with the tracRNA implicated in recruitment of Cas and the crRNA providing specificity for target DNA binding. The resulting Cas9:RNA binary complex could then search the relevant DNA genome. That would include the DNA of the bacteriophage in viral infection or eukaryotic DNA if the CRISPR DNA operon with the genes for Cas 9 and a sg-RNA was transfected into the eukaryotic cell. After RNA binding, the enzyme would change conformation and allow loose DNA binding through Cas 9: PAM interactions.
Studies have shown that the apo form can also bind DNA, but it does so loosely and indiscriminately. It dissociates quickly and binding is affected by generic polyanions such as the glycosoaminoglycan heparin, which indicates its nonspecific nature. Once bound, both off-target and target DNAs would then be surveyed. If a target DNA contained a PAM sequence, the complex would undergo another conformational change to position the HNH and Ruv nuclease catalytic residues and locally unwind the duplex DNA to make the blunt-end cuts.
Cas 9 binding to the PAM site would promote better interaction of the unwound DNA and the bound RNA. If no PAM was present, no catalytically-effective Cas 9:target DNA would form. This prevents off-site cleavage. These allosteric changes and controls are vital to the function of the endonuclease. Here are some findings that support this proposed mechanism:
- the conformation of apo Cas 9 is catalytically inactive;
- on binding RNA to form a binary complex, Cas 9 undergoes a dramatic conformational change, mostly in the REC lobe. However on binding DNA in a nonspecific fashion, the conformational changes are much smaller. This suggests that most changes in conformation occur before DNA binding. In a way, RNA acts as a allosteric activator of the enzyme (as well as the major source of binding specificity to target DNA). Conformational changes can be determined directly by comparison of crystal structures or spectral techniques such as fluorescence resonance energy transfer (FRET) between two different attached fluorophores.
- Cas 9: RNA interactions lead to ordering of the region of the RNA that interacts with the DNA PAM sequence and adjacent deoxynucleotides (a "seed sequence"), allowing the Cas 9:RNA complex to scan and interact with potential DNA targets with PAM sequences;
- Once a PAM site is found, conformational changes leads to unwinding of the dsDNA, which allows heteroduplex formation between the crRNA and the target DNA strand;
- since Cas 9 recognizes a variety of DNA target sequences (but of course only a specific PAM sequence), the binding of the target sequence depends on the geometry, not the sequence, of the target DNA;
- since binding of off-target DNA to the Cas 9:RNA complex occurs but with very infrequent cleavage, binding and cleavage are very distinct steps;
- on specific DNA binding, the HNH catalytic site moves near to the sessile DNA bond site. Crystal structures shown that the active site His is not sufficiently close to facilitate cleavage, suggesting that binding of a second metal ion (see below) may be necessary. Molecular dynamics studies show that the HNH domain is "remarkably plastic".
The animated image below shows the relative conformational changes going from the apo Cas 9 to the binary Cas 9:sgRNA complex to the ternary Cas 9: sgRNA: target DNA complex. The NUC catalytic domain is shown in light blue, the REC (receptor or RNA binding domain) in orange, , sgRNA in red, and target DNA in green. Note again that on binding RNA to form a binary complex, Cas 9 undergoes a dramatic conformational change, mostly in the REC lobe. The pdb protein sequences shown were aligned using pdbEfold.
A potential abbreviated catalytic mechanism for the Ruv nuclease domain is shown in the figure below. The red arrows indicate the second set of electron movements. His 983 acts as a general base to abstract a proton from water making it a more potent nucleophile. An intermediate trigonal bipyramidal phospho-intermediate is formed, which, along with the preceding transition state, is stabilized by the proximal Mg2+ ion (an example of electrostatic or metal ion catalysis). The magnesium is positioned through its interaction with negatively charged carboxyl groups of Asp 10, Glu 762, and Asp 986.
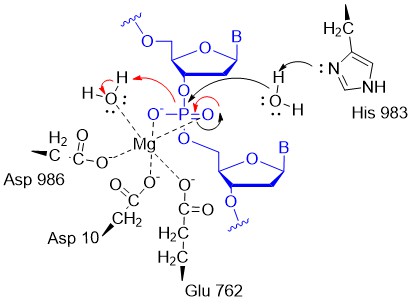
More recent data suggests a second metal ion might be recruited to the Ruv site to further facilitate cleavage of the DNA. The HNH catalytic site has a structure (beta-beta-alpha) and conserved His in common with a class of nucleases that require one metal ion. In contrast, the Ruv catalytic site does not have this common secondary structural motif and has a critical histidine, both common features found in endonucleases that use two metal ions.
CRISPR and Eukaryotic Gene Editing
How could blunt-end cutting of both strands of DNA by Cas 9 lead to the holy grail of specific eukaryotic gene editing with no off-site effects? Cutting the DNA genome seems like a bad idea. It fact, it is potentially so bad that a myriad of DNA repair mechanisms have evolved to fix the cut. These include homologous recombination. If corrective DNA is supplied as well as the components of the CRISPR system, a cell could effectively add the corrective DNA after the double-stranded cut and repair a deleterious mutation. Consult a molecular biology textbook for more insight into homologous recombination.
Mutations in the PAM sequence prevent Cas9 nuclease activity. Hence the NGG PAM sequence is vital for the interactions and activity described above. This would seem to limit the utility of CRISPR-Cas 9 in eukaryotic gene editing, until one realizes that the GG dinucleotide has a 5.2% frequency of occurrence in the human genome, which corresponds to over 160 million occurrences. Even then it might not occur in a desire gene target. Cas 9 nuclease from other bacteria extend the range of activity of the CRISPR/Cas system as they interact with other PAM sequences (NNAGAA and NGGNG for S. thermophilus and NGGNG for N. meningidtis). Likewise, mutations in the S. pyogenes PAM (NGG) have been made as well. A D1135E mutation retains but increases the specificity for the normal NGG PAM site. D1135V, R1335Q and T1337R mutations alter the optimal PAM recognition site to NGAN or NGNG.
CRISPR editing can be easily used to knock out specific genes. In addition, if cells are transfected with a plasmid with many target sequences, the system can be used to edit multiple genes in one experiment. This would be very useful in studies of diseases linked to multiple genes. Since the cost of CRISPR reagents (plasmids, RNAs) is so inexpensive, and the specificity of editing is so high, the great excitement about CRISPR use for gene editing in human disease and for modification of plant and fungal genomes is warranted.
Other systems have been developed to specifically bind to a target DNA sequence and then cleave it. They typically contain a protein that binds to a specific DNA target and an associated endonuclease that cleaves within the target DNA site. Typical prokaryotic restriction enzymes bind to and cut at a specific nucleotide sequence (for example Eco R1 cleaves at G/AATTC palindromic sequences) to form sticky ends. The protein itself binds to this DNA recognition site. Other examples are based on the structure of known transcription factors. Libraries of genetically engineered proteins with Zn finger DNA binding domains (designed for specific DNA target sequences) fused to endonucleases have been created for this purpose. Another example are proteins called TALENs (transcription activator-like effector nucleases). These are fusion proteins containing a TAL effector DNA-binding domain and a nuclease. In each of these cases, a 3D-folded protein is the specific target DNA recognition molecule. Think how much easier it is to make in effect a 1D-DNA recognition element, a simple linear RNA sequence, which would adopt the correct 3D structure on binding of its complementary target.
One major problem in the use of CRISPR for gene editing must be solved: how to get the CRISPR components in the correct cells in an organism. In effect, it's the same problem faced by small drug designers only the components are much larger. Ex vivo applications, when diseased cells are removed from the body, repaired by CRISPR, and then reinjected, are likely to have more success. In these cases, electroporation would allow uptake of Cas 9 and the sg-RNA. In vivo therapy has included use of adeno-associated viruses in which genes for Cas 9 and sg RNA could be encapsulated. This technique, used for other gene delivery systems, has the advantage of being tolerated immunologically. However, this system allows for continual gene expression which is undesirable for gene editing. After an initial "fix" of a mutant gene, continued expression of the CRISPR-Cas 9 genes would increase the chances for off-target cutting. A more recent approach is to delivery the mRNA in artificial lipid nanoparticles that can be taken into cells. Once free and translated into protein and sg RNA inside the cell, gene editing has a chance to occur before the RNA and protein are degraded.
The latest!
Hong Ma et al have corrected the gene for a mutant MYBPC3 gene in an eight cell human embryo that was not transplanted into a uterus. (Hong Ma et al. Correction of a pathogenic gene mutation in human embryos. Nature. doi:10.1038/nature23305. Published online 02 August 2017). The mutation in this autosomal dominant gene causes a type of cardiac myopathy. It can cause sudden death and is found in 1 of 500 people. CRISPR was used to modify the mutant gene. Homologous recombination with the normal allele led to repair of the modified gene.
 Navigation
Navigation
Return to 7B: Methods of Enzyme Catalysis
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 7B: Mechanisms of Enyzme Catalysis

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.