Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 8 - OXIDATION/PHOSPHORYLATION
C: ATP AND OXIDATIVE PHOSPHORYLATION
BIOCHEMISTRY - DR. JAKUBOWSKI
04/15/16
|
Learning Goals/Objectives for Chapter 8C: After class and this reading, students will be able to
|
C1. ATP
Biological oxidation reactions serve two functions, as described in the previous chapter. Oxidation of organic molecules can produce new molecules with different properties. For example, increases in solubility is observed on hydroxylation of aromatic substrates by cytochrome P450. Likewise, amino acids can by oxidized to produce neurotransmitters. Most biological oxidation reactions occur, however, to produce energy to drive thermodynamically unfavored biological processes such as protein and nucleic acid synthesis, or motility. Chemical potential energy is not just released in biological oxidation reactions. Rather, it is transduced into a more useful form of chemical energy in the molecule ATP (adenosine triphosphate). This chapter will discuss the properties that make ATP so useful biologically, and how exergonic biological oxidation reactions are coupled to the synthesis of ATP.
PROPERTIES OF ATP
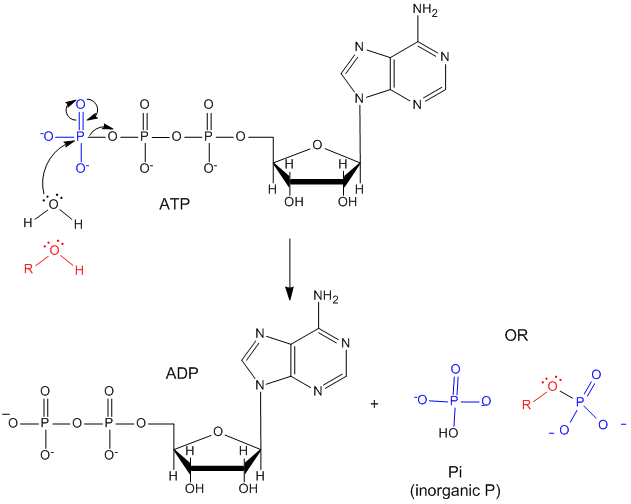
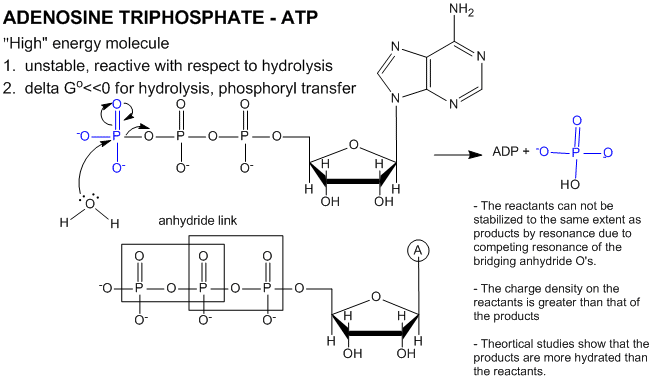
ATP contains two phosphoanhydride bonds (connecting the 3 phosphates together) and one phosphoester bond (connecting a phosphate to the ribose ring). The pKa's for the reactions HATP3- ---> ATP4- + H+ and HADP2- ---> ADP3- + H+ are about 7.0, so the overall charges of ATP and ADP at physiological pH are -3.5 and -2.5, respectively. Each of the phosphorous atoms are highly electrophilic and can react with nucleophiles like the OH of water or an alcohol. As we discussed earlier, anhydrides are thermodynamically more reactive than esters which are more reactive than amides. The large negative ΔGo (-7.5 kcal/mol) for the hydrolysis (a nucleophilic substitution reaction) of one of the phosphoanhydride bonds can be attributed to a relative destabilization of the reactants (ATP and water) and relative stabilization of the products (ADP = Pi). Specifically
- The reactants can not be stabilized to the same extent as products by resonance due to competing resonance of the bridging anhydride O's.
- The charge density on the reactants is greater than that of the products
- Theoretical studies show that the products are more hydrated than the reactants.
The ΔGo for hydrolysis of ATP is dependent on the divalent ion concentration and pH, which affect the the stabilization and the magnitude of the charge states of the reactants and products.
![]() Jmol: Updated
ATP
Jmol14
(Java) |
JSMol (HTML5) | ADP
Jmol14
(Java) |
JSMol (HTML5)
Jmol: Updated
ATP
Jmol14
(Java) |
JSMol (HTML5) | ADP
Jmol14
(Java) |
JSMol (HTML5)
Figure: STRUCTURE AND HYDROLYSIS OF ATP
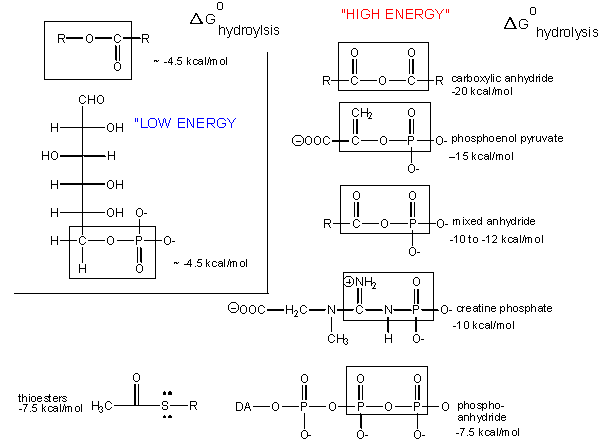
Carboxylic acid anhydrides are even more unstable to hydrolysis than ATP (-20 kcal/mol), followed by mixed anhydrides (-12 kcal/mol), and phosphoric acid anhydrides (-7.5 kcal/mol). These molecules are often termed "high energy" molecules, which is somewhat of a misnomer. They are high energy only in relation to the energy of their cleavage products, such that the reaction proceeds with a large negative ΔGo.
Figure: HIGH ENERGY MOLECULES
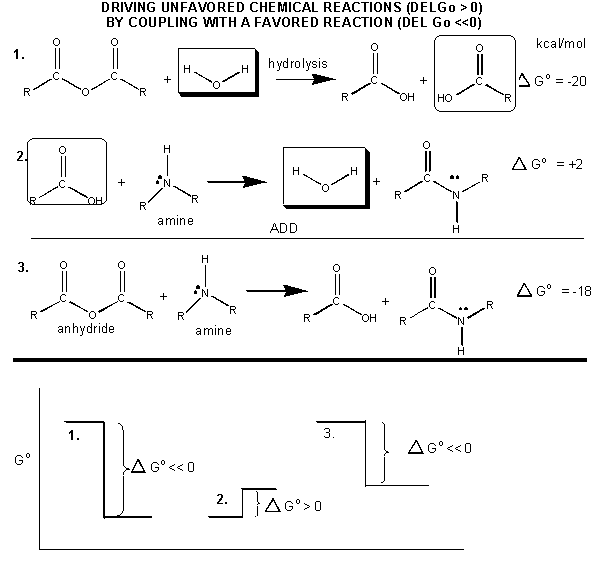
How can ATP be used to drive thermodynamically unfavored reaction? First consider how the hydrolysis of a carboxylic acid anhydride, which has a ΔGo = -12.5 kcal/mol can drive the synthesis of a carboxylic acid amide, with a Δ Go = + 2-3 kcal/mol. The link below shows the net reaction, (anhydride + amine --> amide + carboxylic acid), which can be broken into two reactions: hydrolysis of the anhydride, and the synthesis of the amide.
Figure: MECHANISM: COUPLED SYNTHESIS OF A CARBOXYLIC AMIDE
Now consider the reaction of glucose + Pi to
form glucose-6-P. In this reaction a phosphoester is formed, so the reaction
would proceed with a positive ΔGo
= 3.3. Now if ATP was used to transfer the terminal
(gamma) phosphate to glucose to form Glc-6-P, the reaction proceeds with a
ΔGo = -4 kcal/mol. This
can be calculated since ΔG is a state function and is path
independent. Adding the reactions and the ΔGo's
for glucose + Pi ------> glucose-6-P and
ATP + H2O -----> ADP + Pi gives
the resultant reaction and ΔGo,
glucose + ATP -----> Glucose-6-P + ADP, ΔGo
= -4.
In most biological reactions using ATP, the terminal P of ATP is transferred to a substrate using an enzyme called a kinase. Hence, hexokinase transfers the gamma phosphate from ATP to a hexose sugar. Protein kinase is an enzyme which transfers the gamma phosphate to a protein substrate.
ATP is also used to drive peptide bond (amide) synthesis during protein synthesis. From an energetic point of view, anhydride cleavage can provide the energy for amide bond formation. Peptide bond synthesis is cells is accompanied by cleavage of both phosphoanhydride bonds in ATP in a complicated set of reactions that is catalyzed by ribosomes in the cells. (This topic is considered in depth in molecular biology courses). The figure below is a grossly simplified mechanism of how peptide bond formation can be coupled to ATP cleavage.
Figure: MECHANISM: ATP-DEPENDENT PEPTIDE BOND SYNTHESIS
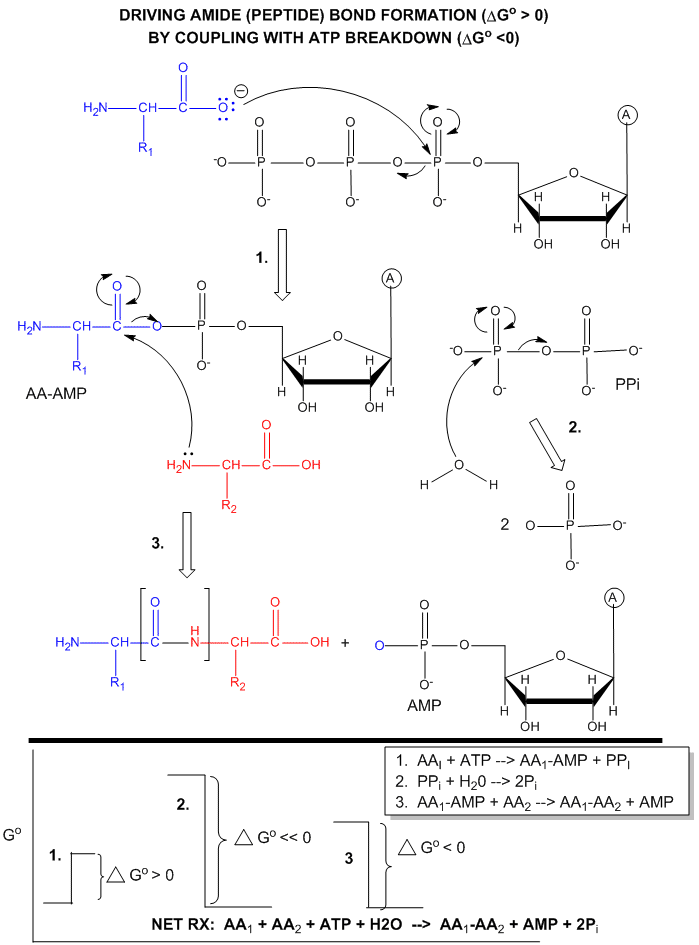
Phosphorylation reactions using ATP are really nucleophilic substitution reactions which proceed through a pentavalent intermediate. The rest of the ATP molecule is then considered the leaving group, which could be theoretically ADP or AMP as well. If water is the nucleophile, the reaction is also a hydrolysis reaction. These reactions are also called phosphoryl transfer reactions.
One last note. ATP exists in cells as just one member of a pool of adenine nucleotides which consists of not only ATP, but also ADP and AMP (along with Pi). These constituents are readily interconvertible. We actually break down an amount of ATP each day equal to about our body weight. Likewise we make about the same amount from the turnover products. When energy is needed, carbohydrates and lipids are oxidized and ATP is produced, which can then be immediately used for motility, biosynthesis, etc. It is very important to realize that although ATP is converted to ADP in a thermodynamically spontaneous process, the process is kinetically slow without an enzyme. Hence ATP is stable in solution. However, its biological half-life is not long since it is used very quickly as described above. This recapitulates a theme we have seen before. Many reactions (like oxidation with dioxygen, denaturation of proteins in nonpolar solvent, and now ATP hydrolysis) are thermodynamically favored but kinetically slow. This kinetic slowness is a necessary but of course insufficient condition, for life.
C2. Coupling of Oxidation and ATP Synthesis under Anaerobic Conditions
Our main goal is to understand how oxidation reactions can lead to ATP synthesis. First let us consider ATP production under anaerobic condition, such as which often occurs during the fight or flight response. You know how terribly you feel when you run a 100 m dash. Your muscles feel horribly due to lactic acid buildup, and you know you can't seem to get enough dioxygen into your body. Under these conditions, a pathway called glycolysis (which you studied in biology) is active. In this pathway, glucose, a 6 carbon hexose, is converted to two, 3C molecules - pyruvate. A detail description is shown below.
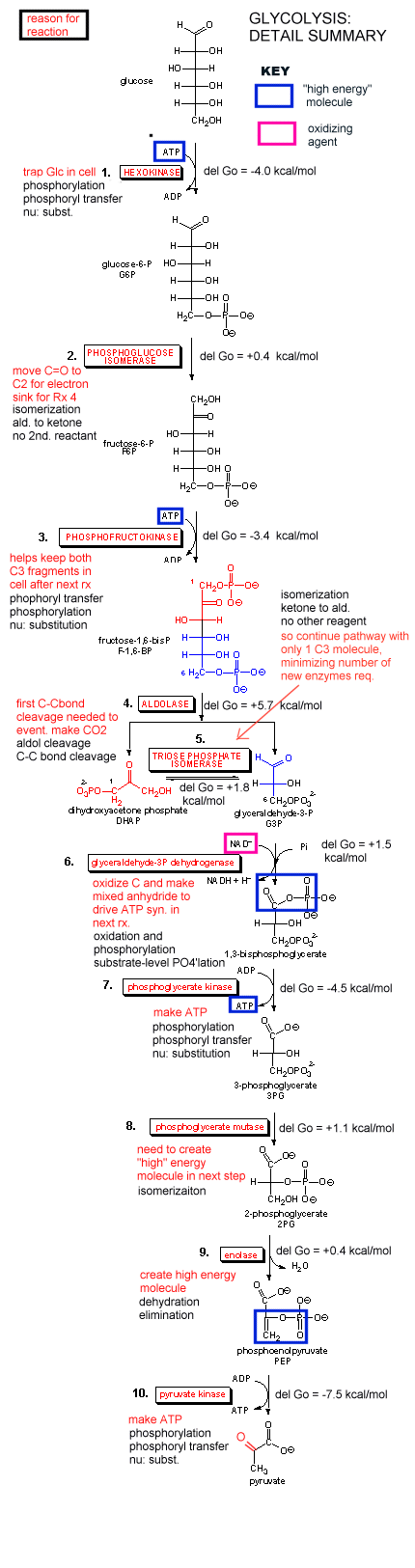
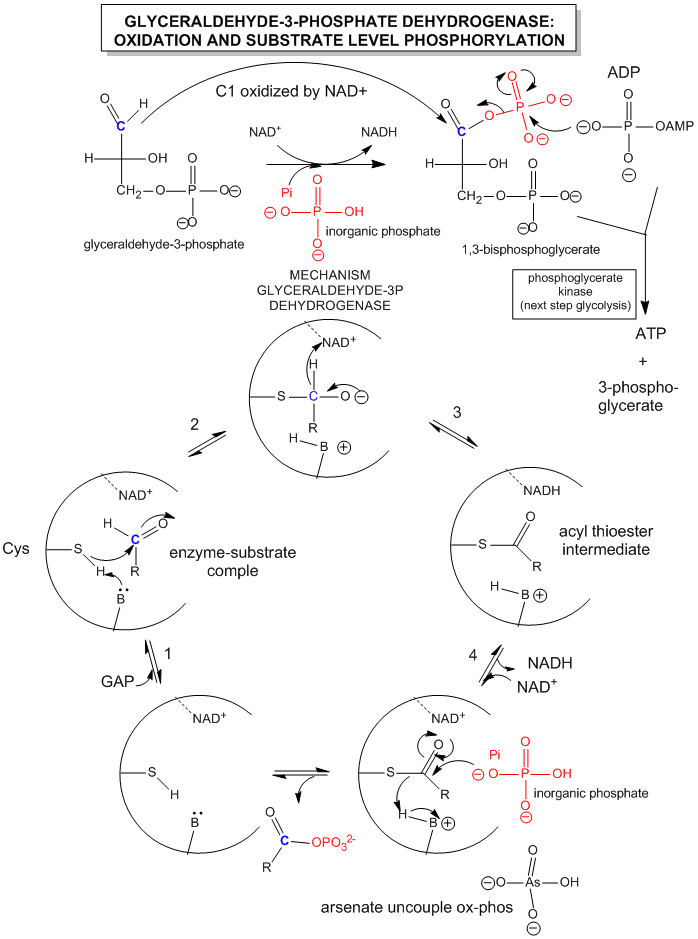
Since we most interested in energy transduction at this point, let's consider just two important step in glycolysis that directly lead to ATP synthesis. Only one oxidative step is found in this pathway, namely the oxidative phosphorylation of the 3C glycolytic intermediate glyceraldehyde-3-phosphate, to 1,3-bisphosphglyercate, a mixed anhydride (see link below for mechanism). The oxidizing agent is NAD+ and the phosphorylating agent is NOT ATP but rather Pi. The enzyme is named glyceraldehyde-3-phosphate dehydrogenase. It contains an active site Cys, which helps explain how the enzyme can be inactivated with a stoichiometric amounts of iodoacetamide. A general base in the enzyme abstracts an H+ from Cys, which attacks the carbonyl C of the glyceraldehyde, forming a tetrahedral intermediate. Instead of the expected reaction (which would be the protonation of the alkoxide in an overall nucleophilic addition reaction at the aldehyde), a hydride leaves from the former carbonyl C to NAD+ in an oxidation step. Notice, this is a two electron oxidation reaction similar to seen in alcohol dehydrogenase. An acyl-thioester intermediate has formed, much like the acyl intermediate that formed in Ser proteases. Next inorganic phosphorous, Pi, attacks the carbonyl C of the intermediate in a nucleophilic substitution reaction to form the mixed anhydride product, 1,3-bisphoshphoglycerate. Although we have formed a mixed anhydride, we cleaved a sulfur ester, which is destabilized with respect to its hydrolysis products (since the reactant, the thioester, is not stabilized by resonance to the extent of regular esters owing to the poor donation of electrons from the larger S to the carbonyl-like C.) In the next step, catalyzed by the enzyme phosphoglycerate kinase, ADP acts an a nucleophile which attacks the mixed anhydride of the 1,3-bisphosphoglyerate to form ATP. Note that the enzyme is name for the reverse reaction. We have coupled oxidation of an organic molecule (glyceraldehyde-3-phosphate) to phosphorylation of ADP through the formation of a "high" energy mixed anhydride, 1,3-bisphosphoglycerate.
The linkage between oxidation of glyceraldehyde-3-phosphate and the phosphorylation of ADP by 1,3-bisphosphoglycerate can be artificially uncoupled by adding arsenate, which has a similar structure as phosphate. The arsenate can form a mixed anhydride at C1 of glyceraldehyde-3-phosphate, but since the bridging O-As bond is longer and not as strong as in the mixed anhydride, it is easily hydrolyzed. This prevents subsequent transfer of phosphate to ADP to form ATP.
![]() Jmol:
Updated Glyceraldehyde-3-phosphate dehydrogenase (NAD)
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Glyceraldehyde-3-phosphate dehydrogenase (NAD)
Jmol14 (Java) |
JSMol (HTML5)
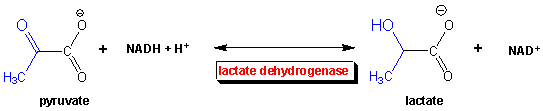
Under anaerobic conditions, glucose is metabolized through glycolysis which converts it to two molecules of pyruvate. Only one oxidation step has been performed when glyceraldehyde 3-phospate is oxidized to 1,3-bisphosphoglycerate. To regenerate NAD+ so glycolysis can continue, pyruvate is reduced to lactate, catalyzed by lactate dehydrogenae. These reactions take place in the cytoplasm of cells actively engaged in anaerobic oxidation of glucose (muscle cells for examples during sprints). Note that the enzyme is named for the reverse reaction, the oxidation of lactate by NAD+.
Lactate in the muscle can go by way of the blood to the liver (where NAD+ is not depleted) and be converted back to pyruvate and eventually back to glucose through a pathway called gluconeogenisis. The liver can export the glucose into the blood from where it can be taken up by the muscle for ATP production. This cycle is called the Cori cycle.
Figure: Cori Cycle
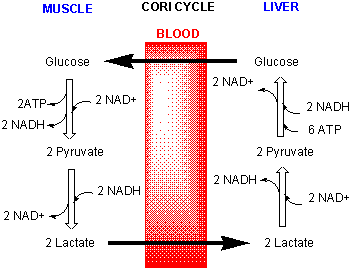
![]() Moodle
Online Quiz (PASSWORD PROTECTED): GLYCOLYSIS
Moodle
Online Quiz (PASSWORD PROTECTED): GLYCOLYSIS
C3. Coupling of Oxidation and Phosphorylation Under Aerobic Conditions
A quick glance reveals that we have taken glucose a small fraction along the way of oxidizing every carbon in it to CO2 and H2O. The complete oxidation happens under aerobic condition when the glycolytic pathway is followed by the Kreb's cycle. Pyruvate formed in glycolysis enters the mitochondrial matrix, and get oxidatively decarboxylated to a 2C molecule, acetylCoA by the enzyme pyruvate dehydrogenase.
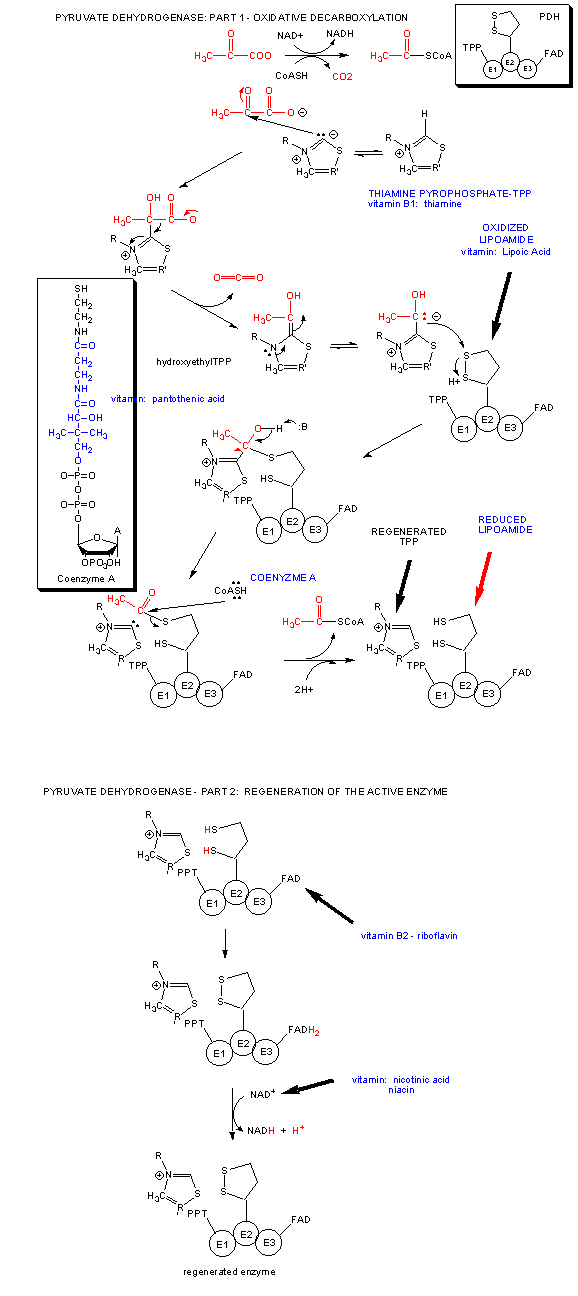
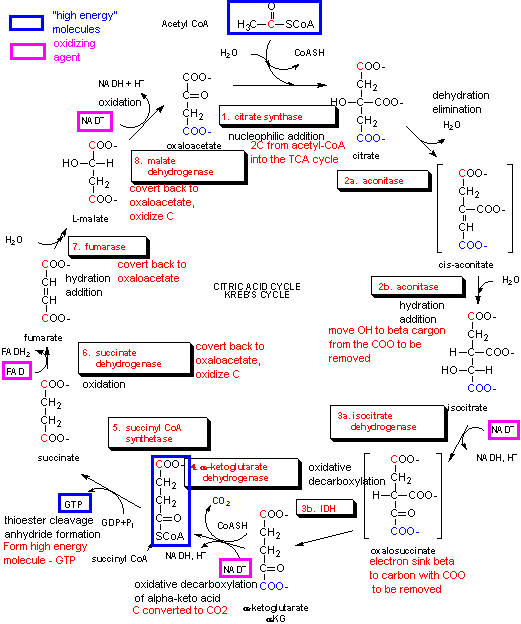
Acetyl CoA then enters the Kreb's cycle (also called the tricarboxylic acid (TCA) cycle. It is shown below.
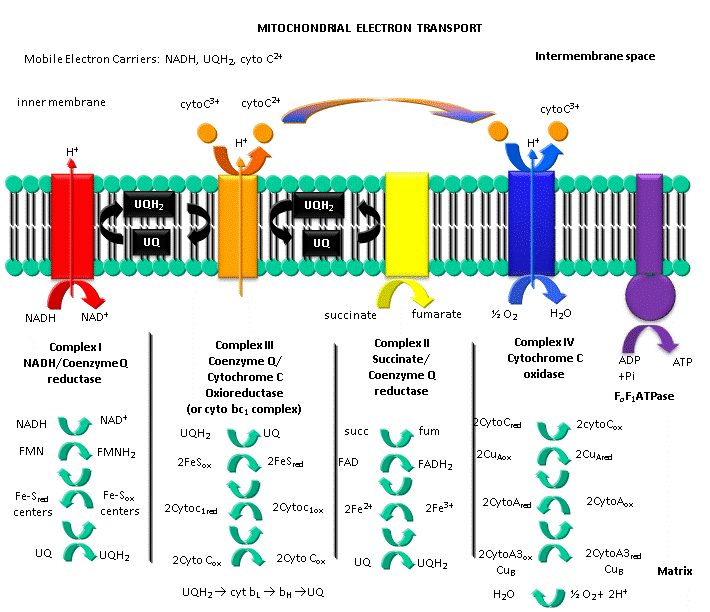
C4. An overview of mitochondrial electron transport
The main oxidizing agent used during aerobic metabolism is NAD+ (although FAD is used in one step) which get converted to NADH. Unless the NAD+ can be regenerated, glycolysis and the Kreb's cycle will grind to a halt. Luckily, under these conditions we are actually continually breathing one of the best oxidizing agents around, dioxygen. NADH is oxidized back to NAD+ not directly by dioxygen, but indirectly as electrons flow from NADH through a series of electron carriers to dioxygen, which gets reduced to water. This process is called electron transport. No atoms of oxygen are incorporated into NADH or any intermediary electron carrier. Hence the enzymes involved in the terminal electron transport step, in which electrons pass to dioxygen, is an oxidase. The enzymes of the Kreb's cycle and electron transport are localized in mitochondria.
Figure: mitochondria
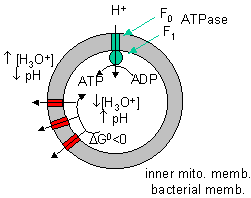
By analogy to the coupling mechanism under anaerobic conditions, it would be useful from a biological perspective if this electron transport from NADH to dioxygen, a thermodynamically favorable reaction (as you calculated in the last study guide - a value of about -55 kcal/mol), were coupled to ATP synthesis. It is! For years scientist tried to find a high energy phosphorylated intermediate, similar to that formed by glyceraldehyde-3-phosphate dehydrogenase in glycolysis, which could drive ATP synthesis (which likewise occurs in the mitochondria). None could be found. A startling hypothesis was put forward by Peter Mitchell, which was proven correct and for which he was awarded the Nobel Prize in Chemistry in 1978. The immediate source of energy to drive ATP synthesis was shown to come not from a phosphorylated intermediate, but a proton gradient across the mitochondrial inner membrane. All the enzymes complexes in electron transport are in the inner membrane of the mitochondria, as opposed to the cytoplasmic enzymes of glycolysis. A pH gradient is formed across the inner membrane occurs in respiring mitochondria. In electron transport, electrons are passed from mobile electron carriers through membrane complexes back to another mobile carrier. Initially, NADH shuttles electrons (2 electron oxidation, characteristic of NAD+/NADH), to a flavin derivative, FMN, covalently attached to Complex I. The reduced form of FMN then passes electrons in single electron steps (characteristic of FAD-like molecules, which can undergo 1 or 2 electrons transfers) through the complex to the lipophilic electron carrier, ubiquinone, UQ.
{kind=link}
{kind=link}
Figure: lipophilic electron carrier, ubiquinone, UQ
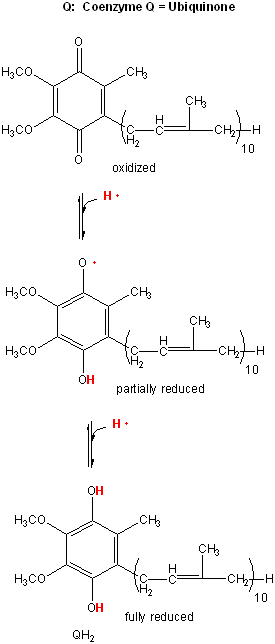
This then passes electrons through Complex III to another mobile electron carrier, a small protein, cytochrome C. Then cytochrome C passes electrons through complex IV, cytochrome C oxidase, to dioxygen to form water. At each step electrons are passed to better and better oxidizing agents, as reflected in their increasing positive standard reduction potential. Hence the oxidation at each complex is thermodynamically favored.
![]() Jmol:
Updated Cytochrome C Oxidase
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Cytochrome C Oxidase
Jmol14 (Java) |
JSMol (HTML5)
Complex II (also called succinate:quinone oxidoreductase) is a Kreb cycle enzyme that catalyzes the oxidation of succinate to fumarate by bound FAD (hence its other name: succinate dehydrogenase). It is not involved in flow of electrons from NADH to dioxygen described above but passes electrons from the reduced succinate to ubiquinone to form fumarate and reduced ubiquone which then can transfer electrons to cytochrome C through Complex III. The crystal structure of this complex has recently been solved by Yankovskaya et al. who have shown that the arrangement of the redox-active sites in the complex minimizes potential oxidation of bound FADH2 by dioxygen, minimizing production of harmful reactive oxygen species like superoxide.
![]() Animation
of electron transport in mitochondria
Animation
of electron transport in mitochondria
![]() Jmol:
Updated Succinate Dehydrogenase (Complex II)
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Succinate Dehydrogenase (Complex II)
Jmol14 (Java) |
JSMol (HTML5)
At each complex, the energy released by the oxidative event is used to drive protons through each complex from the matrix to the intermembrane space of the mitochondria, and is not used to form a high energy mixed anhydride as we saw in the glyceraldehyde-3-phosphate dehydrogenase reaction. The actual mechanism of proton transfer is unclear.
Figure: ELECTRON TRANSPORT AND PROTON GRADIENT FORMATION IN THE MITOCHONDRIA
C5. Complex I (NADH-quinone oxidoreductase)
Now lets explore electron transport in greater detail by looking at the mechanisms of two specific complex, I and IV. Before that look at a detailed view of the entry pathway of electron transport and oxidative phosphorylation.
Figure: Detailed View of Oxidative Phosphorylation (reprinted with permission from Kanehisa Laboratories and the KEGG project: www.kegg.org )
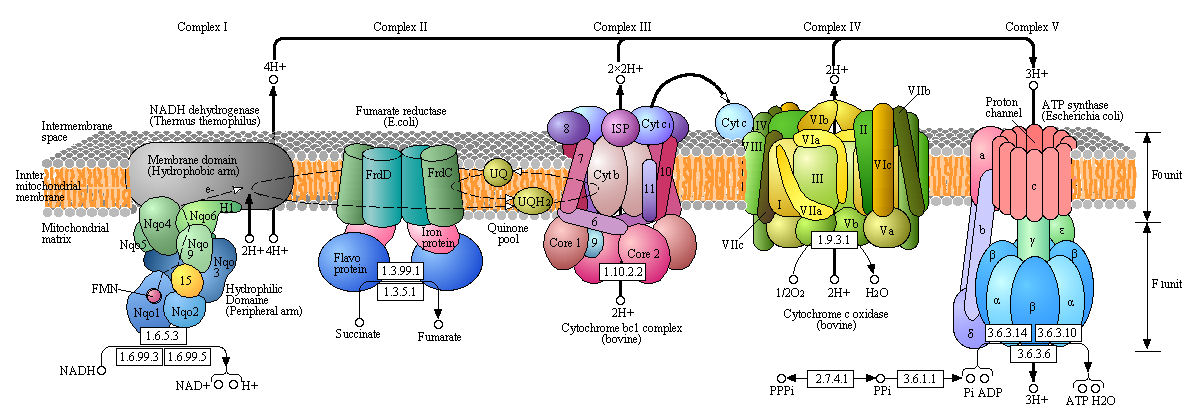
Boxed number represent Enzyme Commission Number. Original KEGG Map with imbedded links.
Complex I - NADH-quinone oxidoreductase
Complex I is located in the inner mitochondrial membrane in eukaryotes and in the plasma membrane of bacteria. Mammalian complex 1 consists of 45 subunits, 7 of which are encoded by mitochondrial gene. Bacteria have only 13-14 subunits. The significance of the extra mammalian subunits is still unclear. A cartoon model and the actual crystal structure of the Thermus thermophilus (bacterial) complex are shown below. The hydrophilic or peripheral domain catalyzes electrons transfer while the membrane domain (encoded by mitochondrial DNA) is involved in active transport of protons.
Figure: Detailed view of Complex I from Thermus Thermophilus
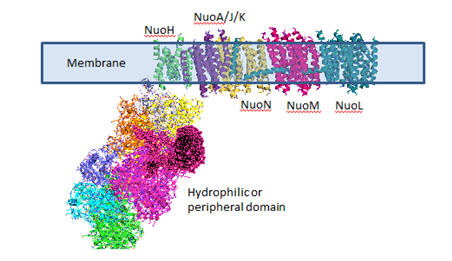
Electron Transport
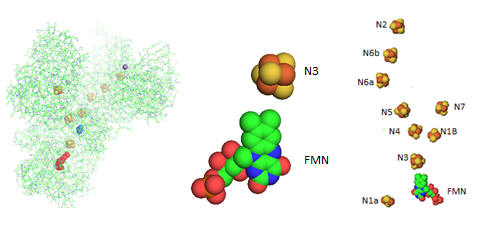
Electron flow occurs from NADH to UQ through a series of one electron carriers in the hydrophilic or peripheral domain of complex 1. Initial handoff of electrons occurs to a flavin cofactor, FMN, and then through a series of Fe/S clusters. In the left figure below, these electron acceptors include a tetranuclear Fe/S cluster (SF4 shown yellow/red spacefill, a binuclear Fe/S cluster (FE2/S2) shown in blue, a FMN flavin mononucleotide shown in red, and a MN (II) ion, shown in purple N1a and N1b are binuclear clusters and N2, N3, N4, N5, and N6 are tetranuclear clusters.
Figure: Electron Flow in Complex I from T. Thermophilus
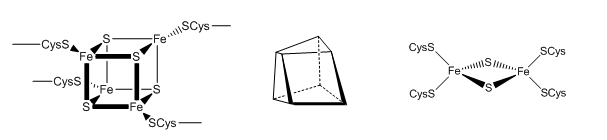
The tetranuclear Fe/S cluster is based on the cubane structure with Fe and S occupying alternating corners of a square in a tetrahedral geometry. Each Fe is also coordinated to thiolate anions. The actual structure is a distorted cube as shown below, along with that of the binuclear cluster, whose bond angle also deviate from those in a tetrahedron.
Figure: Fe/S Clusters in Complex I
Many possible micro-redox states with different standard reduction potential are possible for tetranuclear Fe/S clusters, much as a polyprotic acid has multiple pKa values. The two relevant for Complex I and other tetranuclear clusters are shown below:
a. FeIIFe3IIIS4(CysS)41- + e- ↔ Fe2IIFe2IIIS4(CysS)42- (lower standard reduction potentials)
b. Fe2IIFe2IIIS4(CysS)42- + e- ↔ Fe3IIFeIIIS4(CysS)43- (higher standard reduction potentials)
Electrons are passed singly to oxidized UQ in one electron steps to form UQH2.
Fe-S clusters are synthesized predominately in the mitochondria where they serve as redox cofactors in electron transport as described above. They are ubiquitous in all life forms and serve roles in addition to redox cofactors per se as they serve structural roles in proteins and are used in redox signaling within the cell as they change oxidation states. Many proteins that interact with DNA (repair enzymes, polymerases and helicases) contain an Fe-S cluster.
Evidence suggests that they placed critical roles in the abiotic evolution of life in the absence of oxygen as a a terminal electron acceptor in exergonic oxidation reactions. When oxidation became available, they became potentially became toxic to the cell as the Fe2+ could participate in reactions (such as the Fenton reaction) leading to the generation of deleterious reactive oxygen species (such as superoxide). To prevent toxicity, when delivered to the cytoplasm and nucleus they must carried and delivered by cytoplasmic iron-sulfur assembly (CIA) proteins.
Proton transport occurs in the membrane domain.
Available evidence suggests that 4 protons move from the cytoplasm to the periplasmic space against a concentration gradient during a catalytic cycle of Complex I in bacteria. One appears to be associated with the reduction of UQ at the terminal tetranuclear Fe/S cluster N2. The other three protons move across the membrane domain.
Nqo4 (proximal to the membrane domain as seen in KEGG diagram) residues in chain D have been implicated in H+ flow to the N2 cluster. These include, starting from the N2 cluster, H169, H170, D86, R350, D401, H129, R279, H89, R125, E122, R249, Y257, Y254, Y260, R296 (conserved residues are in bold). The terminal N2 cluster is coordinated by two tandem Cys side chains (C45 and C46) that in their thiolate (deprotonated form) are ligated to the Fe in the cluster. What properties do these amino acids have that make them candidates for this H+ flow?
A model of electron and H+ flow is shown below (after Berrisford and Sazanov, JBC, 284, 29773, 2009). Iron-sulfur clusters N2 and N6b are depicted as O (for oxidized) or R (for reduced). C46 and C45 indicate the tandem cysteines from Nqo6 subunit (nearest the membrane domain). Q/QH2 indicate quinone/quinol. Tyr 87 (Y-O) and Glu 49 (D-O) are proton acceptors. H-path indicates proton delivery pathway from the cytosol to tandem cysteines. One proton from this cycle appears to be transported across the membrane. What happens to the other two protons shown in the diagram?
Figure: Coupled proton and electron transfer at the FeS center in Complex I
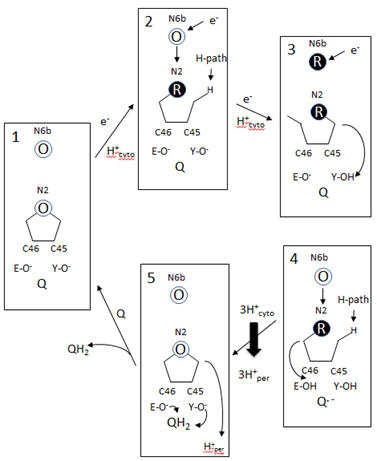
Additional proton are transported by the membrane domain. The NuoL, M, N, A/J/K and H transmembrane domains are shown below. The L, M and N domains have structures similar to proteins involve in the coupled movement of K+/H+ in opposite directions across a membrane (antiporter). There are discontinuous helices in each subunit. Possible residues at the discontinuity buried in the membrane helices are Glu 144 and Lys 234. Would you expect to find these buried in the membrane? If they are part of an unopened channel that is exposed on a concerted conformational change across 3 similar membrane protein domains, how would they participate in proton transfer.
Figure: Transmembrane Domain of Complex I of T. Thermophilus
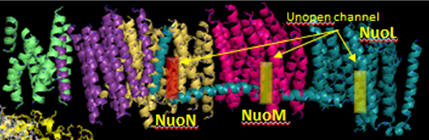
Looking at the left three antiporter subunits L, M, and N, notice a large helix that runs horizontally across all of them. How might that helix function to couple movement of protons across all the antiporter subunits (L, M, and N)? (Hint: think mechanically)
Inhibition of Complex I
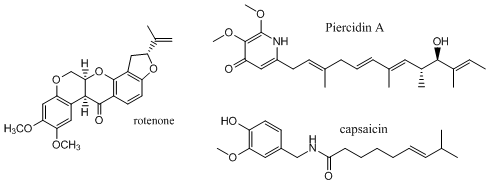
Complex I is inhibited by more than 60 different families of compounds. They include the classic Complex I inhibitor rotenone and many other synthetic insecticides/acaricides. The classes include: Class I/A (the prototype of which is Piericidin A), Class II/B (the prototype of which is Rotenone) and Class C (the prototype of which is Capsaicin). They appear to bind at the same site. From the structure of the 3 prototypes, what are the characteristics of the pharmacophore, the “ideal binding ligand”? Where do they likely bind? How “promiscuous” is the binding site?
Figure: Inhibitors of Complex I
Many devastating neurological diseases are associated with defects in Complex I. In addition to major problems with oxidative ATP production, reactive oxygen species (ROS) increase. The major sites for generation of ROS are Complex 1 and Complex III. Given the locations of the electron carriers at the periphery and internal within the protein complex, which electron carriers might most readily leak electrons to dioxygen? What ROS is likely to form in the process?
Inhibitors might block access of UQ or conformational changes necessary for final reduction of the ubiqinone free radical. Class A inhibitors dramatically increase ROS production. The actual site of ROS production in Complex is a bit controversial. One possible electron donor to dioxygen is FMN. Why is this a likely candidate? Mutants that lack N2 iron-sulfur cluster showed ROS production. Is this consistent with FMN site involvement in ROS production?
In submitochondrial preparations, normal Complex I activity occurs (which leads to formation of a sustained proton gradient). Also reverse electron transport, powered by an artificial proton gradient can occur, which leads to the reduction of NAD+ can occur (see diagram below).
Figure: Normal and Reverse Electron Transport Complex I
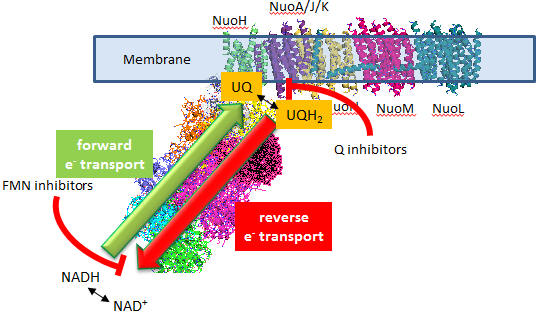
A summary of finding on superoxide production by Complex I is given below:
- Superoxide production is inhibited flavin site inhibitors but not Q site inhibitors.
- Reverse electron transport leads to NAD+ and O2 reduction
- Reverse electron transport superoxide production is inhibited by both flavin and Q site inhibitors
Based on these findings, which site, the flavin or Q site, is involved in superoxide production?
C6. Complex III
TBA
C7. Complex IV - Cytochrome C oxidase (CCOx)
The structure of complex IV is shown in the left figure and to the right in a diagram taken from the KEGG pathways (with permission).
Figure: Cytochrome C Oxidase
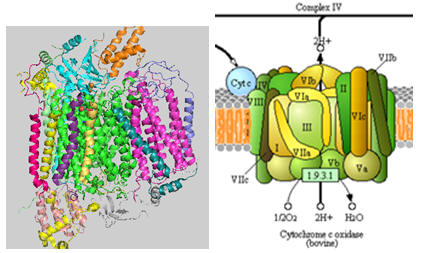
Cytochrome C, the initial “substrate” of this complex, delivers electrons from its heme cofactor to a dinuclear copper cluster, CuA. From there electrons flow to an adjacent heme a (low spin) which transfers them to another heme a3 (high spin) and then finally to dioxygen which is coordinated to the Fe in heme a3 and to an adjacent CuB. The heme a3 Fe:Cu dinuclear cluster is unique among all hemes. Which of the hemes is mostly likely to have two His side chains coordinated to the iron heme? One?
Figure: Heme and Fe:Cu cluster in Cytochrome C Oxidase - 1
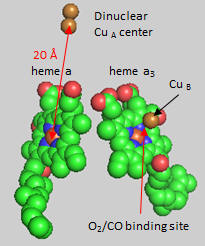
First let’s consider the transfer of electrons from heme a to heme a3 to dioxygen (we will consider entry of electrons into the complex later). What would be the consequence if dioxygen, a substrate for the reaction, dissociated from the heme a3 Fe before it were completely reduced? Suggest a reason for evolution of this key enzyme to have produced the unique heme a3 Fe:Cu dinuclear cluster. Heme a and a3 vary from the heme in hemoglobin as they both have a formyl group replacing a methyl and a hydroxyethylfarnesyl group added to a vinyl substituent. Its structure is shown below. What is its overall charge of the heme in its reduced state? In its oxidized state?
The key challenge has been to understand the redox coupling to H+ transport. How is this done? The formyl group on the heme is coplanar with the heme in both oxidation states. How might this coplanarity effect the charge in the heme?
Figure: Heme-Formyl group of Cytochrome C Oxidase
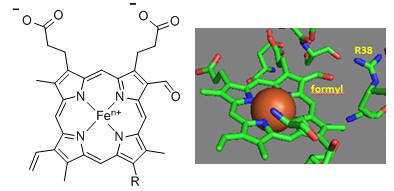
From the figure above, what type of interaction would likely occur between Arg 38 (R38) and the formyl group? What might occur to the protonation state of adjacent protein side chains, specifically Arg 38 (R38) on reduction of the heme? How would this link electron and proton transfer?
How might an electron be “transported” the 20 � distance from the dinuclear CuA cluster to the Fe in heme a and ultimately on to dioxygen? H+ transfer does not occur by physical movement of an individual proton through a “proton pore”. What is the most likely mechanism for transfer? Consider the ratio of the mass of a proton and an electron. Which is more likely to display wavelike behavior?
Crystal structures of oxidized and reduced CCOx show water channels and small “cavities” which calculations show can hold 1-3 water molecules. Hence groups around the heme, including R38 are assessable to water. What might be the function/role of the waters in the channel? Some of the amino acid residues associated with the water channels are shown in the figure below and include R38, S34, T 424, S461, S382, H413. How might these amino acids be involved in proton transfer? Draw an arrow on the diagram showing the direction of proton flow.
Figure: Proton Transfer in Cytochrome C Oxidase
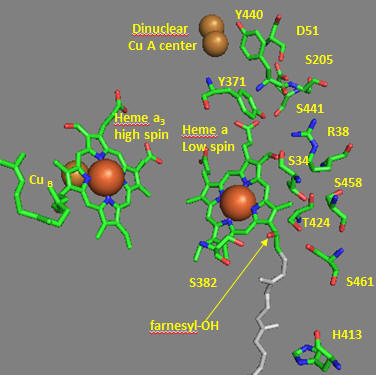
Another consequence of electron transfer to heme a involves the interaction of S382 and the farnesyl OH group, which are close in space and proximal to a water cavity. How might they interact? On reduction of the heme, a conformation change occurs which increases the S382-farnesyl OH group. What effect would this have on the interaction of the two and the S382-L381-Val380 localized conformation? A new water cavity appears to emerge on reduction in this region. How might this impact proton transfer from the matrix?
Now let’s consider the entry site of electrons into the complex and how they might influence proton transport. Structural and functional studies show a key role for Asp 51 (D51) (see figure above). In the oxidized state, D51 interactions with two OH side chains and amide NH backbone groups but is not exposed to water. On reduction, D51 lies on the surface in an aqueous environment. Draw both side chains in their likely protonation state in both the oxidized and reduced complex. What are the consequence of these structures for proton transport?
Near D51 is Y440-S441. The backbone carbonyl group between 440 and 441 forms an “indirect” interaction with R38 which we showed earlier is affected by the redox state of heme a. They are too far apart to form H bonds. Show by adding two waters how an interaction could occur between the carbonyl O and the side chain R38 via Y371. Also show how the water that interacts with Y371 also forms a H bond with the heme a proprionate.
Figure: Role of Amino Acids near D51 in Cytochrome C Oxidase
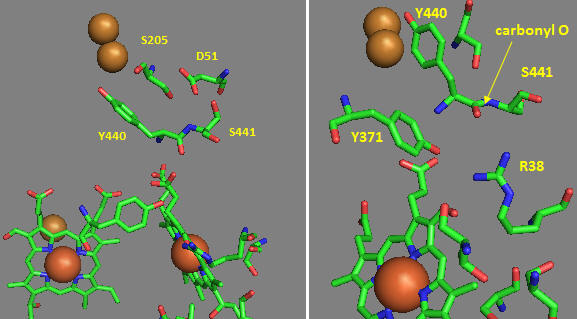
How could these groups participate in a proton transfer mechanism coupled to electron transfer? Would you expect a backbone carbonyl to be involved in proton transfer? Draw a reaction mechanism which shows how protonation of the carbonyl O could lead to formation of an imidic acid which leads to proton transfer from the backbone N. Also show reformation of the normal amide link through a tautomerization reaction. Why would this occur?
How does D51 connect to the this H bond network? In the oxidized state D51 is exposed on the surface. How might it change on reduction? Site specific mutagenesis has been used to change D51. What amino acid replacement might be optimal to affect activity but not protein folding? The mutation uncouples electron and proton transport. Which is likely to be affected?
A final pathway for coupling electron and proton transport has been proposed. Use the information above to complete the following statements: When heme a oxidized, Arg-38 is mainly ____________ (protonated/deprotonated) since _______ is available from the matrix. Asp 51 is ______________ (buried or exposed) and is ______________ (protonated/deprotonated). On reduction of heme a the net charge on heme a _________________ This leads to _________ (increased exposure/decreased exposure) of Asp-51 to the ___________ (intermembrane space, matrix, membrane) and _______ increased/decreased size of the water channel. Hence water molecules are _____________ (taken up/released from) the matrix. Coupled to this, protons on Asp-51 are ___________ (released to or taken up from) the intermembrane space On reoxidaiton of heme a, Asp-51 moves back to the ___________ (interior/exterior) of the protein and the net positive charge on heme a ___________ (increases or decreases) This leads to a _________ (increased or decreased) affinity of the heme formyl group for Arg 38. This leads to proton transfer __________ (to/from) Arg 38 toward Asp 51. Arg-38 then _____________ (gives up/take on) protons from water molecules in the water channel.
C8. Coupling Oxidation and ATP Synthase
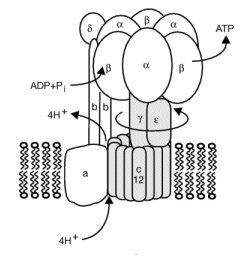
How is this proton gradient coupled to ATP synthesis? Another mitochondrial inner membrane complex, FoF1ATPase, also called ATP synthase or complex V, is found in the inner mitochondrial membrane.
Figure: FoF1ATPase, also called ATP synthase
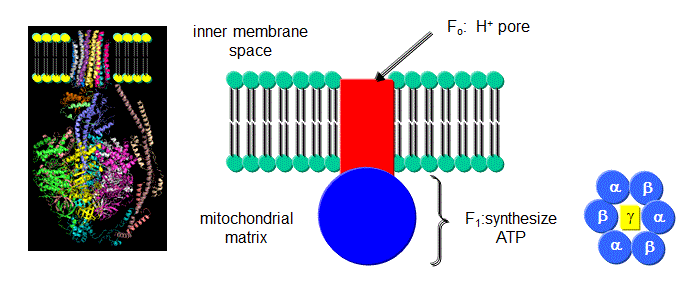
It contains two domains, a transmembrane proton channel, and a enzymatic domain which can either synthesize or hydrolyze ATP. As protons stream through the membrane pore, conformational changes, probably mediated by concerted changes in amino acid side chain pKas. cause the protein to synthesize ATP. Based on kinetic and structural data, Boyer devised an innovative hypothesis for the mechanism of ATP synthesis, which has been verified by structural data. In this model the enzyme, which has multiple subunits, has 3 sites for ATP binding, named L, O, and T. The L or Loose site, binds ATP loosely, the T or Tight site binds it tightly, while the O or Open site does not bind ATP. Although the ΔGo for ATP synthesis in solution is +7.5 kcal/mol, it appears the ΔGo for bound ADP + Pi ----> bound ATP is about 1. Hence the reaction is readily reversible. The difficulty lies in dissociating the bound ATP from the complex. Initially, ADP and Pi bind to the L site. A conformational change occurs, switching the site from L to T, and concomitantly, a T site with ATP bound to an O site which promotes ATP departure. Since the T site has ADP and Pi bound, but has high affinity for ATP, it promotes the synthesis of ATP at that site. This reflects the idea that enzymes bind the transition state (which presumably looks more like ATP than ADP and Pi) more tightly than the substrate. ADP and Pi bind to the newly formed L site which promotes the switch from the T to O site, releasing ATP from the enzyme.
It should now be clear why the enzymes for oxidative phosphorylation in aerobic conditions are membrane bound. Only in this way could a proton gradient be established. Protons must be vectorially transferred in one direction only for a gradient to be established!
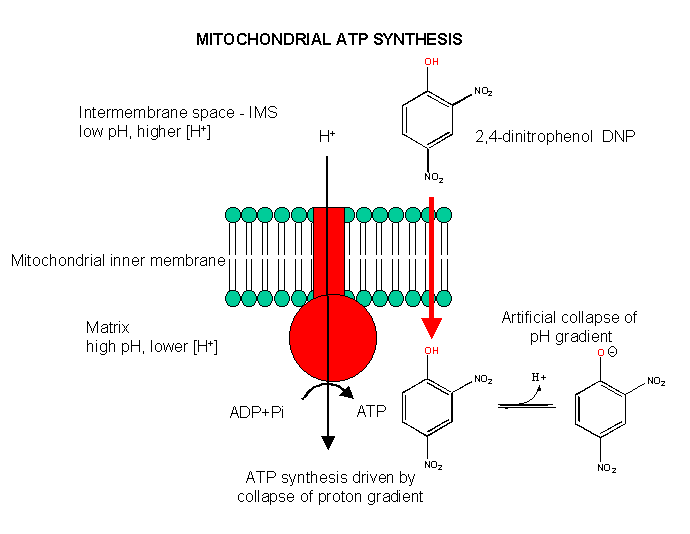
Aerobic ATP production can be uncoupled from electron transfer, as we saw with arsenate uncoupling of ox/phos in aneraobic metabolism in glycolysis. In that case, the energy sources driving ATP synthesis was removed through hydrolysis of the mixed carboxylate/arsenate anhydride. In aerobic metabolism, the energy source is the proton gradient. If this gradient could be artificially collapsed, ATP synthesis would stop, but electron transport (oxidation of NADH through formation of water from dioxygen) would continue. 2,4-dinitrophenol can collapse the proton gradient and act as an uncoupler. In the low pH milieu of the intermembrane space, this weak acid would be protonated. It is also sufficiently nonpolar so as to have reasonable bilayer permeability. When it reaches the higher pH matrix, it can deprotonate. The net effect is to shunt protons through the intermembrane and not through the F0F1ATPase.
Figure: Uncoupling Aerobic Ox/Phos
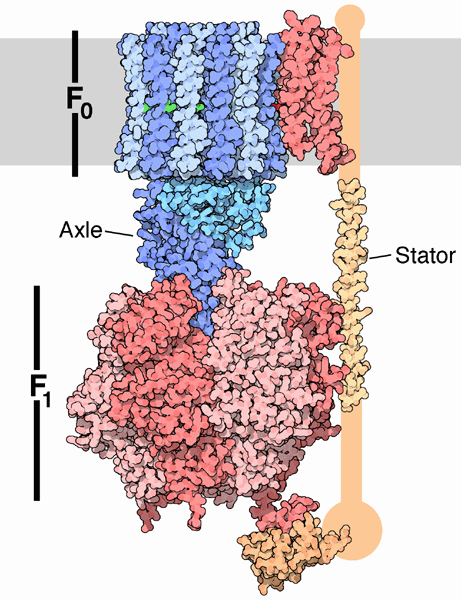
C9. Structural Basis for ATP synthesis
ATP synthase, also called FoF1ATPase, is a rotary motor enzyme. This enzyme is found in the inner membrane of mitochondria, the analogous thylakoid membranes of chloroplasts, and in the cell membrane of bacteria. The enzyme consists of two parts, the membrane bound Fo which is a proton translocator, and the F1 part which has catalytic activity. The enzyme is reversible. If protons flow down a concentration gradient through Fo, ATP is synthesized by F1. Alternatively, ATP hydrolysis by F1 leads to transport of protons through Fo and against a concentration gradient. Isolated F1 can only break down ATP, and not synthesize it. The F1 subunit (with quaternary structure of a3b3 forming a hexagonal ringed structure with a central cavity,occupied by a gamma subunit)is about 80 angstroms from the Fo subunit and both are connected to the rod-shaped γ subunit which spans the center of the a3b3 ring. Energy transduction (necessary to capture the negative free energy change associated with the collapse of the proton gradient to drive the positive free energy change for ATP synthesis) occurs between the two subunits. Noji investigated the structural changes in the γ subunit, wishing to get direct experimental evidence for Boyer's three-state conformational model (L-O-T) for ATP synthesis.
Figure: Boyers three-state conformational model (L-O-T) for ATP synthesis
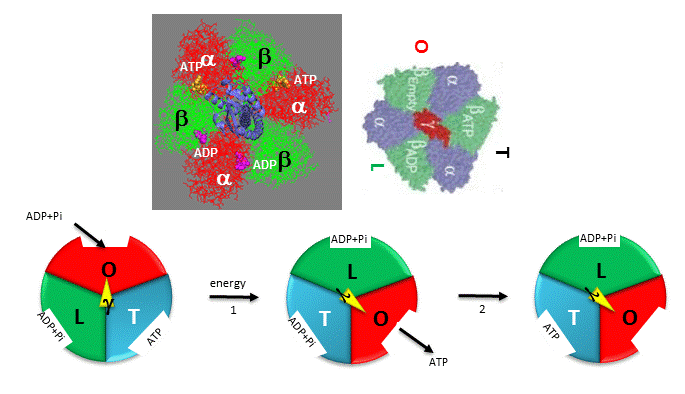
In this model, the F1 subunit exists in three states: an O - open - state with very low affinity for substrates and has no catalytic activity; a L - loose - state with low affinity for substrates and also no catalytic activity, and a T - tight - tight state with high affinity for substrates and with catalytic activity. The F1 subunit consist of three α and three β subunits, which can cycle between three conformations, bind substrate, and have catalytic activity. The collapse of the proton gradient (i.e. the proton-motive force) causes the γ subunit to rotate like a crankshaft relative to the F1 subunit, forcing the β subunit to change conformation from the T to the O (releasing ATP) and then the L (binding ADP and Pi). The γ subunit does not appear to undergo any significant conformational change on ATP hydrolysis as evidenced by tritium exchange studies of amide protons. To prove that the γ subunit rotates, you'd have to observe a single molecule. Since the γ subunit was too small to visually discern its rotation, Noji covalently attached a fluorescein-labeled protein filament called actin to the γ subunit (near where Fo would bind). He then fixed the whole F1 molecule to a glass slip through the a3b3 part, immobilizing that part of the molecule. The γ subunit was free to rotate, which could be detected by observing the fluorescence under a fluorescent microscope from the attached actin filament.
Figure: fluorescein-labeled protein filament called actin to the γ subunit
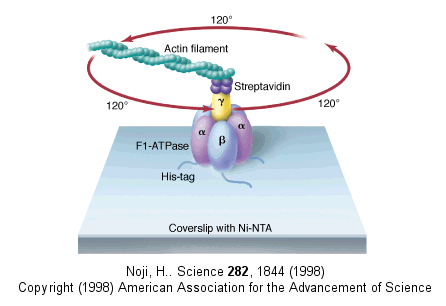
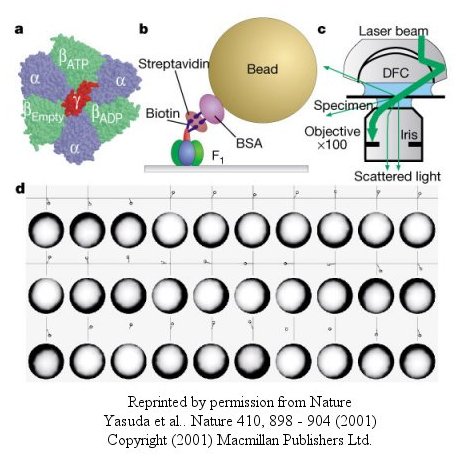
The actin filament rotated only in the presence of ATP. It rotated only counterclockwise, and continued for 10 minutes. This demonstrated that the motion was not random, but a specific motion of the γ subunit. At extremely low concentration of ATP, rotation occurred only in 120o increments, implying one step per molecule of ATP hydrolyzed. (Remember the β subunits are separated by 120o ). As the rotation occurs, there is viscous resistance to movement of the actin filament. He calculate that for a single 120o step caused by hydrolysis of a single ATP molecule, the amount of work was 80 piconewton which is about the free energy of hydrolysis of a single ATP molecule. Incidentally, Boyer was awarded the Noble prize in Chemistry in 1997 for his work. Noji and his colleagues (Nature, 410, 898 (2001), replaced the actin filament with a smaller colloidal gold bead (40 nm diameter) with less frictional resistance to movement and used laser light scattering to probe the rotation of the fixed F1 subunit through the γsubunit.
Figure: smaller colloidal gold bead (40 nm diameter) with less frictional resistance
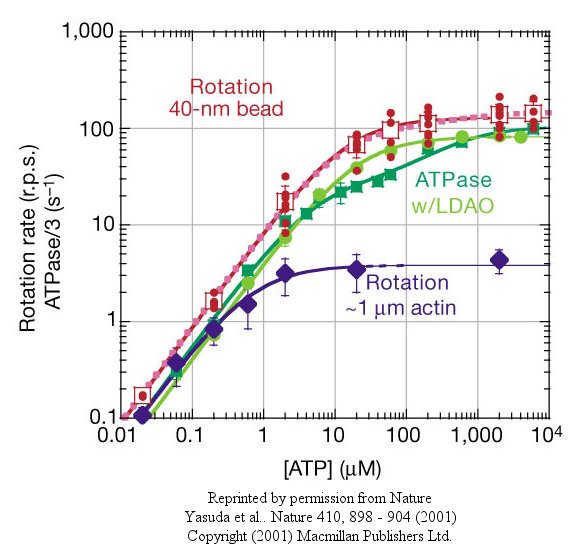
At low [ATP], the motor rotates in 120o steps. At high [ATP], the rotation rate becomes continuous and saturates (with Michaelis/Menten kinetics) at 130 revolutions per second.
Figure: rotation rate becomes continuous and saturates
Recent experiments (Wantanbe, 2010) using immobilized ATPase and magnetic tweezers have addressed the timing of substrate binding and product release when the enzyme is run in reverse (ATP hydrolysis). On rotation of the gamma subunit, the three binding sites change properties. In hydrolysis, ATP binds to the open site, and helps promote the 120 degree rotation. In the next step, ATP is hydrolyzed. In final step, products dissociate. Pi dissociation occurs last from the third site. Hence each of the 3 beta binding sites have different roles. One binds substrate, one performs catalysis, and third releases products. Assuming the synthesis pathway is the reverse of the ATPase reaction, the final release of Pi in ATP cleavage predicts that Pi binds first in the synthetic direction. This would preclude the binding of ATP next which is critical since its concentration during synthesis can be 10x higher than that of ADP. As Pi is bound first, only ADP, not ATP can bind next.
The gamma subunit rotation plays a "catalytic" role as its rotation induces cyclic conformational changes in the beta subunit of the synthase. Can ATP synthesis occur without the gamma subunit by a mechanisms which involves a less proficient, but concerted set of cyclic change in beta subunit conformation? Apparently it can. Uchihashi et al have used high speed atomic force microscopy (AFM) to study the (alpha-beta)3 hexagon from the F1 subunit without the gamma subunit. They found that upon ATP hydrolysis, the beta subunits underwent conformational changes in the same counterclockwise rotary direction as when the gamma subunit was present.
Figure: AFM Study of Conformational Changes in F1 "gammaless" subunit
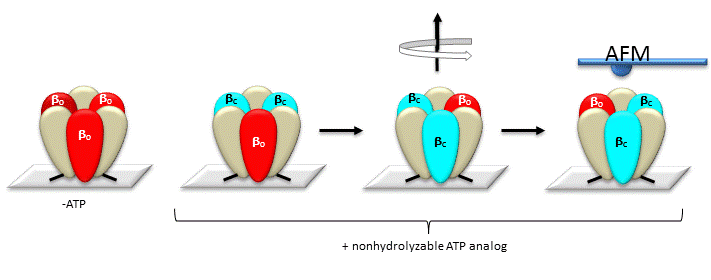
These experiments conclusively show that the F1 subunit is effectively a rotary motor with the gamma subunit acting as a rotor in the stationary hexagon ring composed of the 3 pairs of alpha/beta subunits which acts as the stator (stationary part of an electric rotary motor).
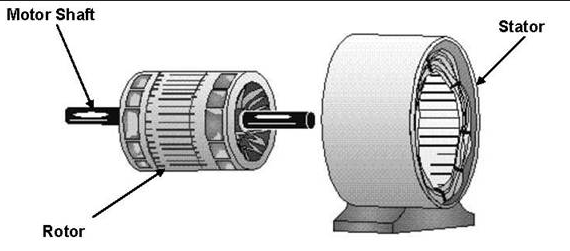
thttp://www.powerpulse.net/techPaper.php?paperID=142&page=3
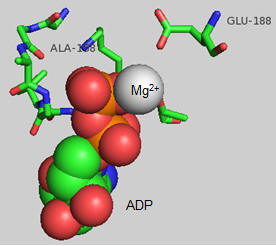
The actual amino acids involved in the mechanism of ATP synthesis/hydrolysis are still not clearly defined but Glu 190 on the beta subunit clearly acts as a general base. The figure below shows bound ADP and the proximity of Glu 188. Ala 158 is thought to move towards the active site after a conformational change, with the nonpolar methyl side chain displacing an adjacent water molecule which could leave as a product of ATP synthesis.
C10. How does the pH Gradient collapse lead to ATP synthesis?
The mechanism by which the proton gradient drives ATP synthesis involves a complex coupling of the F0 and F1 subunits. A more detailed image of the whole ATO synthase complex is shown below.
Figure: Detailed View of F0F1 ATP synthase structure
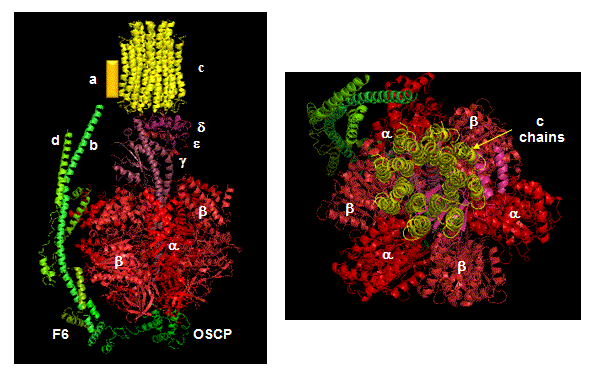
Closer views of the c subunits and a yellow rectangle representing the a subunit (missing in the combined crystal structure) comprise the Fo part of the complex are shown below. These subunits reside in the inner membrane of the mitochondria (or cell membrane of bacteria) and are involved in proton transport from matrix (or cytoplasm of a bacteria) to the inner membrane space (or periplasmic space of bacteria). The multiple c subunits consist of two very hydrophobic helices connected by a loop in a helix-loop-helix motif.
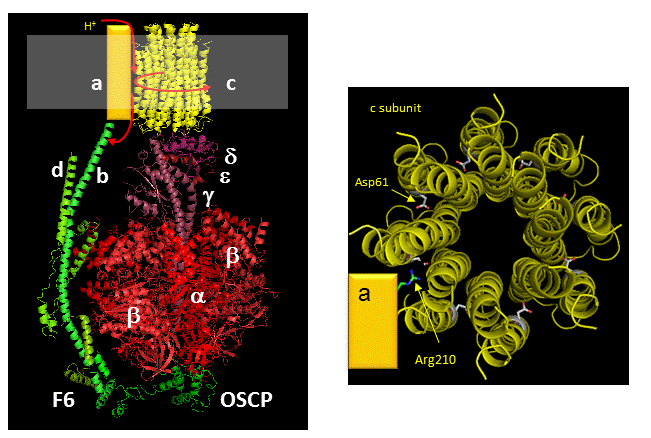
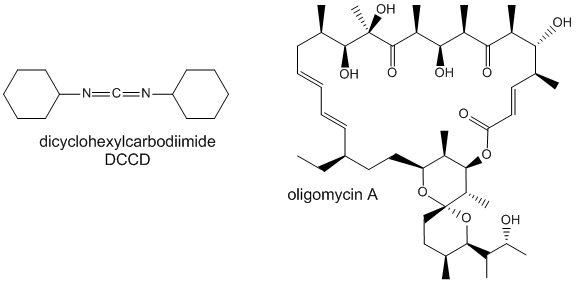
Two classic inhibitors (structures shown below) of ATP synthase interact with the Fo subunit. One, oligomycin A, binds between the a and c subunits and blocks proton transport activity of the Fo subunit. Oligomycin A sensitivity requires, paradoxically, OSCP (Oligomycin-Sensitivity Conferring Protein which is analogous to the bacterial delta subunit), a stalk protein subunit distal to Fo which couples Fo and F1. Another inhibitor, dicyclohexylcarbodiimide reacts with a protonated Asp 61 in c subunits of F0. It does so even at pH 8.0 which indicates that the pKa of the Asp 61 is much higher than usual. This might occur if the Asp is a very hydrophobic environment. The modification of one As 61 in only one c subunit is necessary to stop Fo activity. The protonated carboxyl group donates a proton to a N atom in DCCD, which then reacts with the deprotonated Asp to form an O-acyl isourea derivative.
Figure: Structure of Oligomycin A and DCCD - Inhibitors of proton transport by Fo
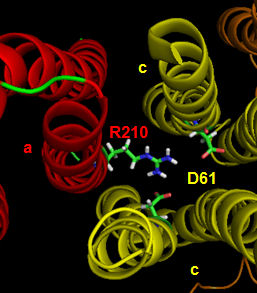
The figures below shown the structure of the ac complex from E. Coli. Protons flow to the a chain Arg 210 which is between two Asp 61 on adjacent c chains. One of the Asp 61 is protonated allowing it to alter conformation and essentially rachet in the membrane domain in a motional faciliated by the development of a neutral protonated Asp.
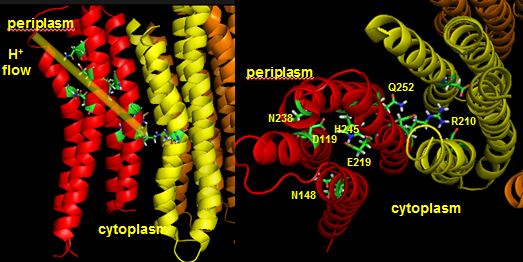
Protons from the inner membrane space or in the periplasmic space (in the above figures) then flow from the periplasm by forming a "handshaking" proton transfer relay which delivers another proton to the deprotonated Arg 210 allowing the circular ratching of the c subunits in the membrane to continue. A set of polar residues entirely within subunit a, including Gln 252, Asn 214, Asn 148, Asp 119, His 245, Glu 219, Ser 144 and Asn 238 provide the path as illustrated below.
When a proton is passed to the unprotonated Asp 61, a conformational change in the protonated c subunit occurs. This leads to changes in c subunit interactions which seems to ratchet the c12 core. Since the c12 oligomer contacts the γsubunit connecting the Fo stalk and F1 ATPase units, the γ subunit rotates, leading to sequential conformational changes in each of the 3 contacted (αβ)2 dimers of the F1 enzyme. This leads to changes in ATP affinity through cycling each through the L, O, and T conformations.
Figure: Coupling Proton Flow in F0 to Conformation Change
Reprinted by permission of Nature. Rastogi & Girvin. Nature 402, 263-268 (1999) Copyright 1999 McMilllan Publishers LTD
![]() animation 2: ATP synthesis mechanism
animation 2: ATP synthesis mechanism
![]() Molecular
Movies (scroll down to ATP synthase)
Molecular
Movies (scroll down to ATP synthase)
![]() Mitochondria:
Harvard
Mitochondria:
Harvard
![]() Powering the
Cell: Mitochondria
Powering the
Cell: Mitochondria
![]() XVIVO: Scientific Animations
XVIVO: Scientific Animations
![]() ATPase
animations from the MRC
ATPase
animations from the MRC
In summary, FOF1ATPase (or synthase) is a rotary enzyme that ultimately couples collapse of a proton gradient (a chemical potential gradient which contributes to the transmembrane electrical potential) to a chemical (phosphorylation) step. The rotor, which is in contact with both the FO proton pore, and the F1 synthase, moves with respect to both subunits, which couples them. Motion of course is relative so the rotor can be thought of as static with the FO and F1 subunits as rotating. The FO pore can hence to be considered an electrical motor and the F1 synthase a chemical motor. Carrying the analogy of a motor even further, the FO electrical motor turns the F1 chemical motor into a generator, not of electricity but of ATP. The figure and link below, taken from the Protein Data Bank, go into more depth about this nanomotor.
C11. CAN A PROTON GRADIENT SUPPLY ENOUGH ENERGY FOR ATP SYNTHESIS?
Experimental evidence shows that it can. The FoF1ATPase complex can be removed from membranes and placed in a liposome into which ADP and Pi have been encapsulated. The pH of the outside of the vesicles is then lowered several pH units. Under these circumstances, ATP is generated inside the vesicle proving that a gradient alone can drive its synthesis.
Mathematical analyses show that it can as well. Consider a typical pH gradient (-1.4 pH units) across the inner membrane of respiring mitochondria (with the outside having a lower pH than inside making the inside more depleted in protons). Clearly there is a chemical potential difference in protons across the membrane. However, another factor determines the thermodynamic driving force for proton translocation across the membrane. A transmembrane potential exists across the inner membrane of the mitochondria, as it does across most membranes. The source of the membrane potential will be discussed in signal transduction chapter. The inside is more negative than the outside, giving the membrane a transmembrane electrical potential. of about -0.14 V. Clearly, protons would be attracted to the other side of the membrane (into the matrix) by this potential difference, which then augments the chemical potential difference as well. A simple mathematical derivation shows that indeed, a proton gradient can supply enough energy for ATP synthesis, especially when coupled to a transmembrane electrical potential.
Figure: A simple mathematical derivation
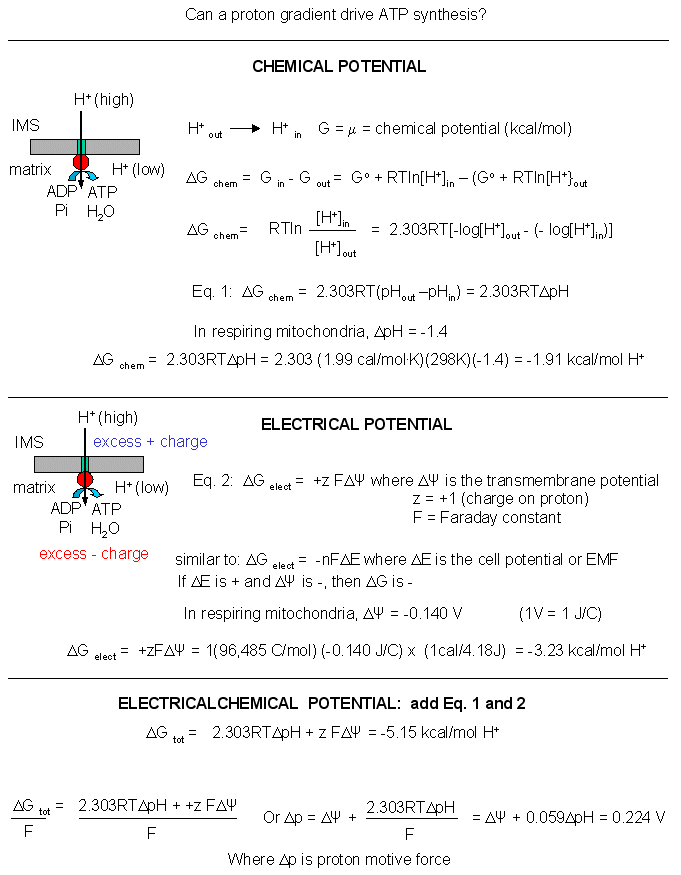
The sum of the electrical and chemical potentials are called the electrochemical potential, which when divided by nF gives the proton motive force.
Note: In the above discussion, we dealt with two different proton translocating methods:
Figure: two different proton translocating methods
- Complex I, III, and IV, which couple uphill proton movement (from the higher pH matrix to the lower pH intermembrane space) to oxidation (NADH + O2 to NAD+ + H2O).
- Downhill movement of protons through F0F1 ATPase which couples to ATP synthesis by the enzyme.
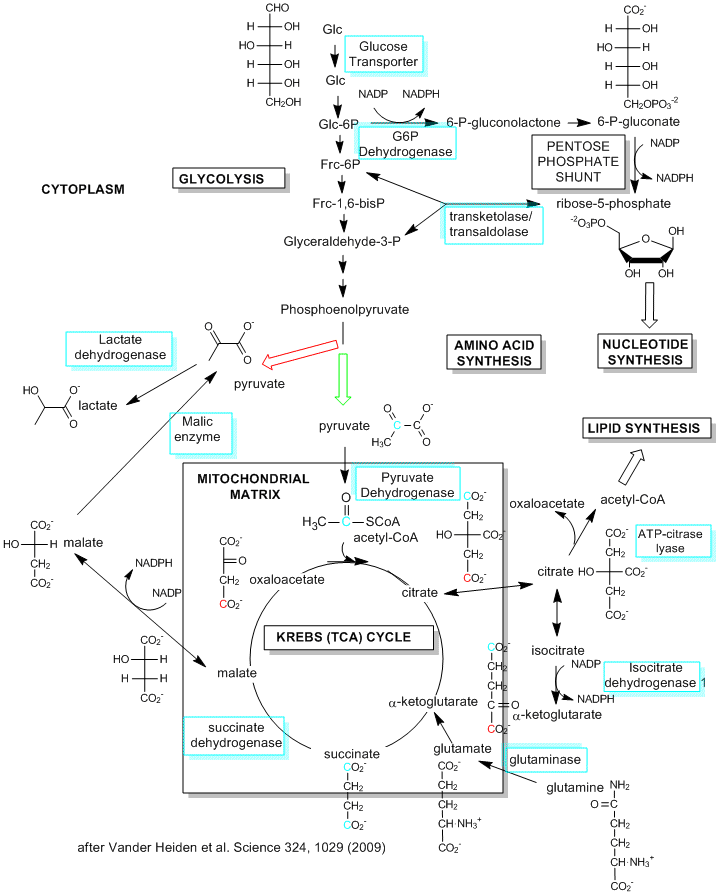
C12. A Comparisoin of Metabolic Needs in Nonproliferating and Proliferating Cells
Although most of this chapter deals with the extraction of energy from glucose through the production of ATP in glycolysis (anaerobic) and the TCA cycle and oxidative phosphorylation (aerobic) in the mitochondria, cells have other needs that must be met, especially the need for reductive biosynthesis to produce fatty acids, proteins, and nucleic acids. Which need prevails? Vander Heiden et al, in a recent review, suggest that it depends on the metabolic needs of the cell. Our understanding of metabolic pathways and their control derive mostly from the study of nondividing cells that are terminally differentiated. In these cells, the need for reductive biosynthesis is minimal so cells extract energy most efficiently from glucose through aerobic oxidation of the glycolytic end product, pyruvate, through mitochondrial oxidative phosphorylation. What about cells that are actually dividing and differentiating? They argue that these cells have a great need for reductive biosynthesis (think of the need to duplicate the contents of the entire cell, which effectively increases the biomass). This would mandate that cells maximize the production of small molecule precursors for synthesis of larger molecules. These small molecule precursors include acetyl CoA for the synthesis of fatty acids for membranes, glucose-6-phosphate for the synthesis of ribose and deoxyribose for the synthesis of RNA and DNA, and a myriad of small molecules or additional metabolites arising from glycolysis and the TCA cycle. If these small molecules, which are produced in the cytoplasm or move from the mitochondria to the cytoplasm, are removed from the energy producing pathway for reductive biosynthesis, how does the cell meet its energy needs?
One type of proliferating cell that has been well studied is a tumor cell. These cells, which have great need for reductive biosynthesis, have long been know to undergo aerobic glycolysis, and in the process produce large amounts of lactate. This effect was observed by Warburg in 1956, who thought the effect arose through defective mitochondria in tumor cells (which is not the case). Proliferating, single cell organisms also engage in aerobic glycolysis. It now appears that proliferating, non-tumor cells from multicellular organisms do as well. Aerobic glycolysis (glycolysis in the presence or absence of dioxygen) would obviously occur most readily under adequate nutritive conditions which would be present in multicellular organisms receiving a constant stream of nutrients delivered by the blood. Under restrictive nutrient conditions, cells would undergo cell phase arrest to minimize proliferation. In contrast, differentiated and nonproliferating cells from multicellular organisms, with no need for significant reductive biosynthesis, would obtain energy most efficiently through mitochondria aerobic pathways.
A little stoichiometry will clarify the differing needs required by the pathway which leads to the most efficient use of glucose for ATP production (through converison to pyruvate and its continued conversion of carbon dioxide aerobocially) and use of glucose for reductive synthesis of the palmitic acid (16:0). The net equation for the production of 16:0 from metabolites of glucose (acetyl CoA formed from progression of glucose to pyruvate in glycolysis and its oxidative subsequent oxidative decarboxylation to acteyl CoA in the mitochondria) is:
8 CH3(CO)SCoA + 7 ATP + 14 NADPH --> 1 palmitic acid + 7 ADP + 7 Pi + 14 NADP+
(Note: NADPH is a phosphorylated version of NADH found in the cytoplasm and is used for reductive biosynthesis instead of NADH. Reduced nicotinamide adenine dinucleotide molecules needed for reductive biosynthesis are differentiated from those produced during catabolism by being phosphorylated and by being predominantly found in a different cellular compartment (the cytoplasm compared to the mitochonria).
From where do all the needed ATP and NADPH molecules derive? Lets consider that question starting from glucose.
-
1 Gluose produces around 36 ATPs from mitochondrial TCA cycle followed by oxidative phosphorylation.. Hence on a molar ratio basis, 0.2 mol mol of glucose are required to produce the necessary 7 mol ATP for synthesis of 1 mol of 16:0. .
-
1 Glucose produces 2 NADPH when the first product in glycolysis, glucose-5-phosphate is withdraw from glycolysis and moved into an altenrative pahway, the pentose phosphate shunt, that produces ribose 5-phosphate for RNA and DNA production. Hence 7 mol glucose (35x the amount needed to make the required ATP) are needed to produce the required 14 mol of NADPH required to produce 1 mol of 16:0.
-
4 Glucose molecules produce 8, 2C-CH3(CO)SCoA (with the other 8 carbons atoms originally in 8 pyruvates lost as CO2 on conversion to acetylCoA by pyruvate dehydrogenase).
Hence there is greater molar need for glucose to be use for production of the small molecule intermediates necessary for 16:0 reductive bioynthesis in proliferating cells than the molar need for glucose to produce the energy (in the form of ATP) required for 16:0 synthesis. This suggests that targeting important but to some "boring" enzymes involved in glycolysis and the TCA cycle (which to reiterate are the sources of the small molecule precursors for reductive biosynthesis) might inhibit tumor proliferation.
Proliferating cells have high ATP/ADP and NADH/NAD ratios which leads to feedback inhibition of important steps in energy production, including the synthesis of citrate from oxaloacetate and pyruvate in the first step of the TCA cycle. Under these conditions, citrate leaves the mitochondria where it cleaved in an ATP depended fashion back into acetyl CoA (which can now be used for fatty acid synthesis) and oxaloacetate. To allow continue TCA activity, glutamine, an amino acid which is also metabolized in high quantities in proliferating cells, can be converted in the mitochondria to glutamic acid which after loss of ammonia forms alpha-ketoglutarate, an intermediate in the TCA cycle. This allows continued energy production in the TCA cycle.
How much of glucose is used for energy production versus small molecule precursor productions. Vander Heiden at al suggest that in proliferating and tumor cells, about 85% is used in lactate production, and 5% used in mitochondrial oxidation, while 10% is shunted for precursor production. 60% of glutamine is also used (as described above) for biosynthesis. It would appear that lactate that results from this process is wasteful of carbon atoms that could go into reductive biosynthesis. However, it can be recycled through the Cori cycle, in which the liver converts it into glucose which is exported.
Figure: Metabolic Pathways in Proliferating Cells
C131. Feeding and Fasting: The Regulation of storage and breakdown of glucose and lipids - The role of PPARs
We have spend little time discussing the detailed anabolic and catabolic pathways of metabolism. That is the topic of a another biochemistry course. However, it should be clear that the one pathway should be activated and the other inhibited, depending on the energy state of the individual. In the well fed state (high levels of carbohydrates and lipids), glycogen, triacylglycerides, and fatty acids synthesis should be activated, while glycogen breakdown (glycogenolysis), mobilization of triglycerides reserves (breakdown of TAGs to form free fatty acids), and fatty acid oxidation should be minimized. In the fasting state, the opposite pathways should be activated. The regulatory control of these opposing processes is complicated but PPARs have been shown to have a major role. PPARs (peroxisome proliferator-activated receptors) are nuclear receptors that are ligand-gated transcription factors. These proteins were initially discovered to be the binding target of small synthetic drugs called peroxisome proliferators. Later the relevant physiological ligands were found to include long chain polyunsaturated fatty acids, oxidized fatty acids, and eicosonoid derivatives of arachidonic acid (20:4Δ5,8,11,14). PPAR, in the presence of ligand binds a second protein, the retinoid X receptor (RXR) which binds 9-cis-retinoic acid). The heterodimer binds to peroxisome proliferator response element in the promoter region of genes involved in lipid transport and metabolism, and activates their transcription. Given these facts, common chronic diseases with lipid abnormalities (cardiovascular disease, diabetes, obesity) would be expected to be affected by PPARs. There are three types of PPARs: α, β, and γ. Only the major two types, α and γ, will be discussed.
| PPAR Type | location | ligand activator | effects |
| α | brown adipose tissue, liver (some in kidney, heart, and skeletal muscle | long chain unsaturated fatty acid like linolenic acid, oxidized fatty acid, eicosanoids (8S-HETE, LT B4 | fatty acid catabolism - FA transport, FA oxidation in peroxisomes and mitochondria, |
| γ | adipose cells, some in colon | 15-deoxy-D-prostaglandin J2 | storage of fatty acids - lipoprotein lipase, adipocyte FA binding protein, FA transport; acyl CoA synthase |
Fatty acids are oxidized when food is scarce, but are stored as triacylglycerides when they are abundant. PPARs α and γ have differential effects in the fed and fasting states:
| Organ | Fed: Synthesize FA, triacylglycerides. CHO and fat in circulation Increased PPAR | Fasting: oxidize FA, break down triacylglycerides Increased PPAR |
| Liver (PPAR-α) |
Glc taken up by liver where it can be stored as glycogen. If glycogen reserves are high, Glc is funneled through glycolysis to pyruvate then to acetyl-CoA. Acetyl CoA then is used in the synthesis of fatty acid, which are esterifed to glycerol to form TAGs. These leave liver as VLDL (very low density lipoprotein). Sterol response element binding protein (SREBP) levels increase, leading to increase in transcription of genes involved in above processes. |
FAs oxidized to Acetyl-CoA. Ketone bodies increase. Stimulated by increased expression of PPAR-α in fasting state. Increased FAs in liver (headed toward oxidation) might bind to PPAR-α and increase its activity. (αβcosθ) |
| Adipocyte (PPAR-γ) |
SREBP and PPAR-γ levels increases (from insulin signaling). Also SREBP activates PPAR-γgene transcription. Lead to uptake of Glc and FA into fat cells (through stimulation of breakdown of blood TAGs, to fatty acids which can be imported into fat cells. Glc through glycolysis to glyceraldehyde 3P which with FAs are converted to TAGs. Increased TAGs lead to leptin release by adipocytes. (This hormone leads to decreasing storage of TAGs. _(AG) | SREBP and PPAR-γ levels lows. TAGs converted to glycerol and FA, mostly for export; Some however reesterifies from FA and glycerol made reverse of glycolytic pathway, called gluconeogenesis; Transcription of an important enzyme in this pathway, PEPCK, is activated under control of PPAR-γ |
Drugs that bind to and either mimic (agonist) PPAR -αorγ effects are useful therapeutically in conditions characterized by lipid abnormalities (diabetes, cardiovascular disease). Drugs that bind to and activate PPAR-g (Rezulin, Avandia) can lower blood glucose levels and are used to treat type II diabetes. Drugs that activate PPAR-a (fibrates like gemfibrozil) can lower serum triglycerides (by stimulating liver fatty acid oxidation). Both drugs ultimately lower serum lipids.
PPARs also have an effect on plasma lipoprotein (LDL, HDL) levels. Both also might have a role in inflammation, which can promote cardiovascular disease. Fibrates, which interact with PPAR-a , appear to inhibit the inflammatory response mediated by the immune system by decreasing the release of protein "hormones" or cytokines, from stimulated immune cells.
![]() Pathways
for PPAR-mediated activation of gene transcription.
Pathways
for PPAR-mediated activation of gene transcription.
![]() Pathway
for PPAR-αmediated activation of gene transcription
Pathway
for PPAR-αmediated activation of gene transcription
C14. Links and References
- Nature 465 (2010) 441-447. doi:10.1038/nature09066
- Nature 465 (2010) 428-429.
- Journal Biological Chemistry 284, (2009) 29773–29783.
- Journal Biological Chemistry 286 (2011) 18056–18065
- Wantanbe, R. et al. Nature Chemical Biology, 6, 814-820 (2010)
- Vander Heiden, M. et al. Understanding the Warburg Effect: The metabolic requirements of cell proliferation. Science 234, pg 1029 (2009).
- Kersten, S. et al. Roles of PPARs in health and disease. Nature. 405, pg 421 (2000)
- Yankovskaya, V. et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 299, pg 700, 671 (2003)
- Oliver, S. Demand Management in Cells. Nature. 48, pg 33 (2002)
- Rastogi and Girvin. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 402, pg 263 (1999)
- Larsen et al. Dietary Advice on Q and Extension of Life-Span in C. Elegans by a Diet Lacking Coenzyme Q (free radicals and aging?). Science. 295, pg 54, 120 (2002)
- Lower et al. How Bacteria Respire minerals. Science. 292. pg 1312, 1360 (2001)
- Echtay et. al. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 408, pg 609 (2000)
- Chen et al. Atomically defined mechanism for proton transfer to a buried redox center in a protein. Nature. 405, pg 814 (2000)
- Echtay et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 415. pg 96 (2002)

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.