Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 5 - BINDING
D: BINDING AND THE
CONTROL OF GENE
TRANSCRIPTION
BIOCHEMISTRY - DR. JAKUBOWSKI
Last Updated: 03/30/16
|
Learning Goals/Objectives for Chapter 5D: After class and this reading, students will be able to
|
D1. Introduction to Transcription
Imagine you have been given a string approximately 2 meters long, which represents an unwound, deproteinated, human chromosome. In actuality, such naked DNA does not exist in the cell nucleus. Rather, it is wound around a series of positively charged histone proteins. In the electron microscope it resembles beads on a string. This structure is wound into a cylindrical "solenoid" structure which is further packaged to fit into the nucleus along with the rest of the chromosomes. There just happens to be a small dot in the dead center of the string you have been given. It represents the gene for a particular protein called metallothionein. This gene is expressed and the protein metallothionein is made when cells are exposed to heavy metals like Cd. How might the cell not express the gene, or express it to a small, constitutive level, in the absence of Cd exposure? How might the gene might be activated - through transcription of the gene and translation of the resulting mRNA to form the metallothionein protein upon exposure to Cd. (Hint: Cd doe not DIRECTLY bind to DNA.)?
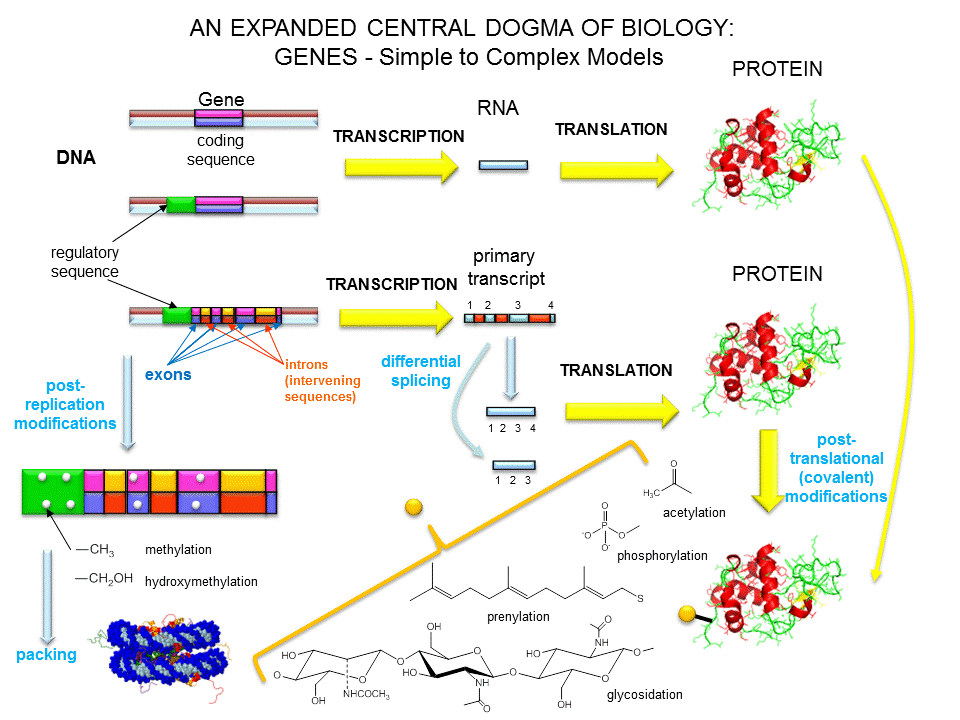
One of the central questions of modern biology is what controls gene expression. As we have previously described, genes must be "turned on" at the right time, in the right cell. To a first approximation, all the cells in an organism contains the same DNA (with the exception of germ cells and immune cells). Cell type is determined by what genes are expressed at a given time. Likewise, cell can change (differentiate) into different types of cells by altering the expression of genes. The Central Dogma of Biology describes how genes are first transcribed to messenger RNA (mRNA), and then the mRNA is translated into a corresponding protein sequence. The links below should be reviewed by those who have little background on the Central Dogma of Biology and of the nature of a gene.
- overview of Central Dogma of Biology for the non-biochemistry major
- A view of genes and their products: Simplicity to Complexity
![]() Updated
Simple DNA Tutorial
Jmol14 (Java) |
JSMol (HTML5)
Updated
Simple DNA Tutorial
Jmol14 (Java) |
JSMol (HTML5)
Once made, proteins can then be post-translationally modified, localized to certain sites within the cells, and ultimately degraded. If functional proteins are considered the end-product of gene expression, the control of gene expression could theoretically occur at any of these steps in the process.
Figure: PROCESSES THAT AFFECT THE STEADY STATE CONCENTRATION OF A PROTEIN
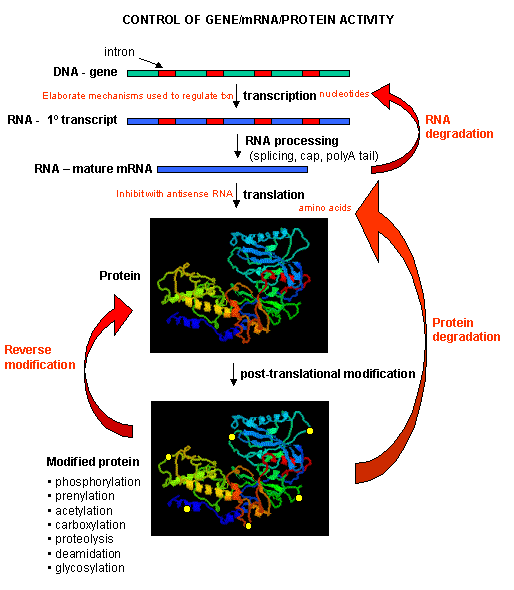
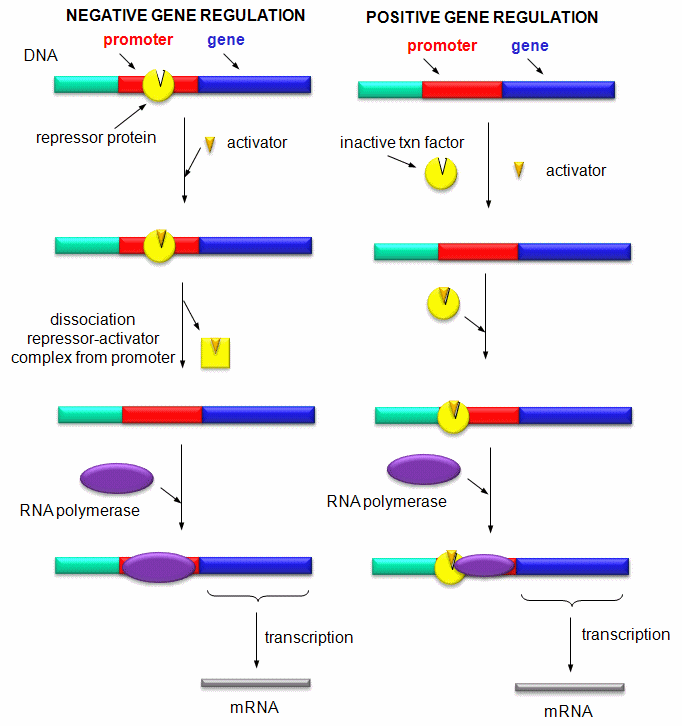
Mostly, however, gene expression appears to be controlled at the level of transcription. This makes great biological sense, since it would be less energetically wasteful to induce or inhibit the ultimate expression of a functional protein at a step early in the process. How can gene expression be regulated at the transcriptional level? Many examples have been documented. The main control is typically exerted at the level of RNA polymerase binding just upstream (5') of a site for transcriptional initiation. Other factors, called transcription factors (which are usually proteins), bind to the same region and promote the binding of RNA polymerase at its binding site, called the promoter. Proteins can also bind to sites on DNA (operator in prokaryotes) and inhibit the assembly of the transcription complex and hence transcription. Regulation of gene transcription then becomes a matter of binding the appropriate transcription factors and RNA polymerase to the appropriate region at the start site for gene transcription. Regulation of gene expression by proteins can be either positive or negative. Regulation in prokaryotes is usually negative while it is positive in eukaryotes.
Figure: Positive and Negative Regulation of Gene Transcription
D2. Control of Transcription in Prokaryotes
The regulation of the genes involved in lactose utilization won Jacob and Monod (of MWC fame) the Nobel Prize. Lactose can be used as the sole source of carbon by E. Coli. Three genes are required for lactose utilization, beta-galactosidase (lac Z, cleaves lactose to Gal and Glc), galactoside permease (lac Y, transports Lac into the cell) and thiogalactoside transacetylase (lac A, function unknown). These genes follow one another on the DNA, and have 1 promoter region. On transcription and translation, one long poly-protein is made, which is cleaved post-translationally to form the individual proteins.
Figure: FUNCTION OF PROTEINS IN GALACTOSE UTILIZATION
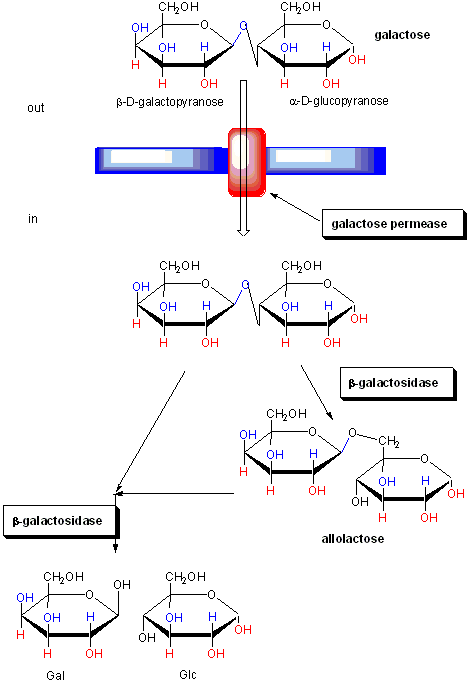
In addition, another gene, the Gal repressor, is found just upstream of the Gal utilization genes. It has its own promoter (PI). A gene cluster, including promoter and any regulatory DNA sequences is called an operon, for example, the Lac operon. In this case, transcription from the operon is induced in response to a molecular signal - i.e. the presence of lactose, or allolactose. The signal binds to the repressor protein, which is bound to the operator DNA, which in the absence of the signal inhibits transcription. When the signal, in this case allolactose or another beta-galactosides, such as isopropylthiogalactoside (IPTG), binds to the repressor protein, a conformation change occurs in the repressor, resulting in a higher Kd for the operator DNA, and subsequent dissociation of the repressor-galactoside complex. Transcription ensues.
Figure: IPTG and Lactose Structures
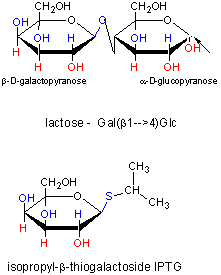
IPTG is an inducer of the lac operon but is not a substrate for the enzymes produced.
Figure: INDUCTION OF LAC OPERON
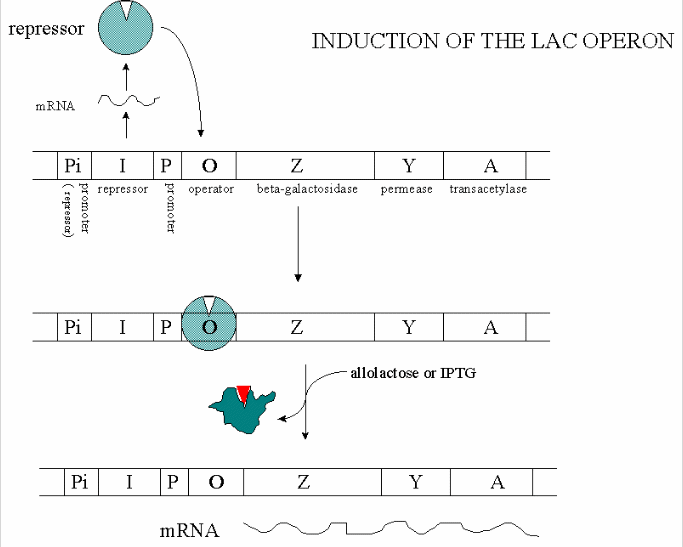
Many analogous but distinct methods are used to control gene transcription in prokaryotes. The control of lac operon transcription is but one example.
D3. Control of Gene Transcription in Eukaryotes
Three major differences exists in the control mechanisms used to regulate gene transcription in eukaryotes compared to prokaryotes.
-
multiple changes occur in the structure of chromatin at the site of transcription
-
positive mechanisms regulate transcriptions much more often than negative ones.
-
transcription and translation occur at spatially and temporally distinct sites and times.
The genomes of eukaryotes are much larger than prokaryotes. This poses some problems with respect to binding. Remember, DNA binding proteins demonstrate both nonspecific and specific binding. Nonspecific binding may help a protein find a specific site in the genome, but as the size of the genome increases, the chance of finding multiple specific sites randomly distributed increases. This problem can be avoided if multiple proteins are required to generate an active transcription complex. The chance of finding two or more specific sites for different proteins in proximity at sites other than required for gene transcription are very low. Multiple negative regulators would not be needed since just the binding of one regulator would probably be sufficient. Most eukaryotic genes have about 5 regulatory sites for binding transcription factors and RNA polymerase. Examples of these transcription factors are show in the figure below.
Figure:
![]() Control
of globin gene transcription
Control
of globin gene transcription
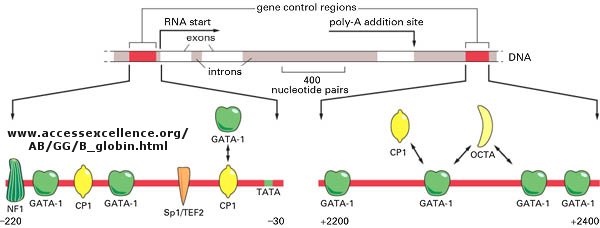
Figure:
![]() Example
of transcription complexes
Example
of transcription complexes
(reprinted with permission from Kanehisa Laboratories and the KEGG project: www.kegg.org )
![]()
Light can even regulate gene expression by indirectly activating an inactive transcription factor in plants. The transcription factor PIF3 binds to a promoter region (G-box) of light-responsive genes. Only when PIF3 binds another transcription factor, Pr, is transcription activated. Pr, a "photoreceptor" is found in an inactive form in the cytoplasm. When it absorbs red light, it undergoes a conformational shift and moves into the nucleus, where it can bind PIF3 and activate transcription. The activation complex is inactivated when Pr interacts with far-red light.
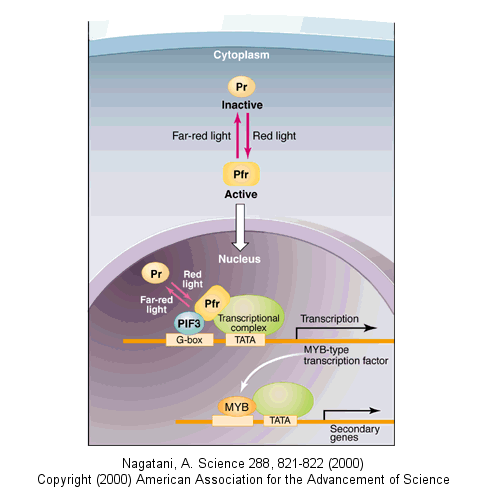
![]() animation:
p53 and gene transcription from the HHMI (loads slowly)
animation:
p53 and gene transcription from the HHMI (loads slowly)
D4. DNA Binding Sites
Since RNA polymerase must interact at the promoter site of all genes, you might expect that all genes would have a similar nucleotide sequence in the promoter region. This is found to be true for both prokaryotic and eukaryotic genes. You would expect, however, that all transcription factors would not have identical DNA binding sequences. The sequences of DNA just upstream of the start site of the gene that binds protein (RNA polymerase, transcription factors, etc) are called promoters. The table below shows the common DNA sequence motif called the Pribnow or TATA box found at around -10 base pairs upstream from the start site, and another at -35. Proteins bind to these sites and facilitate binding of RNA polymerase, leading to gene transcription.
Prokaryotic Promoter Sequences
|
Promoter |
-35 Region |
Spacer | -10 Region | Spacer | RNA start |
| trp operon | TTGACA | N17 | TTAACT | N7 | A |
| tRNAtyr | TTACA | N16 | TATGAT | N7 | A |
| λP2 | TTGACA | N17 | GATACT | N6 | G |
| lac operon | TTTACA | N17 | TATGTT | N6 | A |
| rec A | TTGATA | N16 | TATAAT | N7 | A |
| lex A | TTCCAA | N17 | TATACT | N6 | A |
| T7A3 | TTGACA | N17 | TACGAT | N7 | A |
| consensus | TTGACA | TATAAT |
A similar sequence is found in eukaryotes (consensus TATAAA) located about 25 nucleotides upstream from the transcription start site. It is called the Goldstein-Hogness box.
![]() Jmol:
Updated TATA Box Binding Protein
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated TATA Box Binding Protein
Jmol14 (Java) |
JSMol (HTML5)
In addition, in eukaryotes, sequences further upstream called response elements bind specific proteins (such as CREB or cyclic AMP response element binding protein) to further control gene transcription.
Eukaryotic Response Elements (RE)s
|
Regulatory agent |
Module |
Consensus | DNA bound | Factor | Size (daltons) |
| Heat Shock | HSE | CNNGAANNTCCNNG | 27 bp | HSTF | 93,000 |
| Glucocorticoid | GRE | TGGTACAAATGTTCT | 20 bp | Receptor | 94,000 |
| Cadmium | MRE | CGNCCCGGNCNC | . | ? | . |
| Phorbol Ester | TRE | TGACTCA | 22 bp | AP1 | 39,000 |
| Serum | SRE | CCATATTAGG | 20 bp | SRF | 52,000 |
| Antioxidant | ARE | GTGACTCAGC | |||
| Pheromone (fungus) | ACAAAGGGA | ||||
| Hypoxia | HRE |
CCACAGTGCATACGT GGGCTCCAACAGGTC CTCTCCCTCCCATGCA |
Hypoxia Inducible Factor | 826 aa | |
| Peroxisome Proliferator Activated Receptor (PPAR) | PPRE | aGG_CAAAGGT(CG)A | PPAR | 59,000 | |
| Steroid (general) (progesterone, androgen, mineralcorticoids, glucocorticoids | AGAACAxxxACAAGA (inverted repeat) |
![]() Eukaryotic
Promoter Database (EPD) - annotated non-redundant collection of
eukaryotic POL II promoters, for which the transcription start site has been
determined experimentally. Link to
article
describing EPD and its use.
Eukaryotic
Promoter Database (EPD) - annotated non-redundant collection of
eukaryotic POL II promoters, for which the transcription start site has been
determined experimentally. Link to
article
describing EPD and its use.
In addition to promoters, there are other DNA sequences that are more distal to the transcription start site than the proximal promoter that affect gene transcription. These include enhancers, silencers and insulators. The figure below shows how an enhancer might affect gene transcription by bringing distal DNA (perhaps thousands of base pairs away from the start site of transcription) close to the promoter for a gene. One could easily imagine how a silencer would work in an analogous fashion.
Figure: Enhancers of Transcription
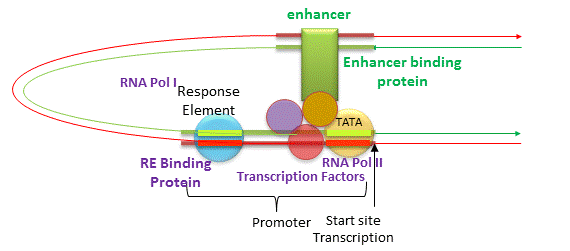
Insulators are needed to prevent an enhancer (or silencer) from activating (or inhibiting) another nearby gene that should be affected. They are sequences between the enhancer (or silencer) and the promoter or a gene or a cluster of genes. Eukaryotic insulators have a CCCTC nucleotide site which bind proteins called CCCTC binding factors (CTCF).
The following figure and captions, reprinted from Nature with permission (as shown in the figure) shows a more detailed description of a simple eukaryotic transcriptional unit that controls gene transcription.
Figure: Eukaryotic promoters and regulatory regions
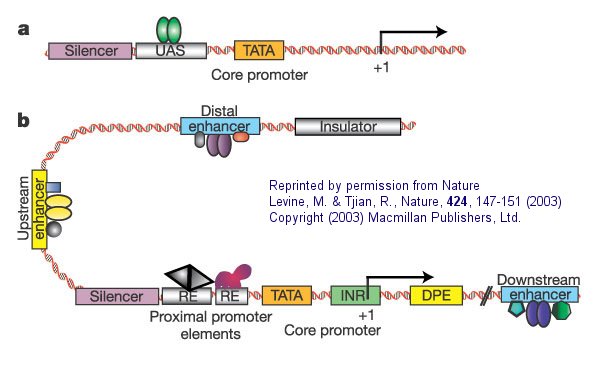
a. "Simple eukaryotic transcriptional unit. A simple core promoter (TATA), upstream activator sequence (UAS) and silencer element spaced within 100�200 bp of the TATA box that is typically found in unicellular eukaryotes".
b. "Complex metazoan (belonging to the kingdom Animalia) transcriptional control modules. A complex arrangement of multiple clustered enhancer modules interspersed with silencer and insulator elements which can be located 10�50 kb either upstream or downstream of a composite core promoter containing TATA box (TATA), Initiator sequences (INR), and downstream promoter elements (DPE)".
The following figure and caption, again taken with permission from Nature, shows a more detailed view of a multi-subunit general transcription apparatus, including tissue-specific and gene-selective subunits and transcription initiation complexes.
Figure: Eukaryotic multisubunit general transcription apparatus
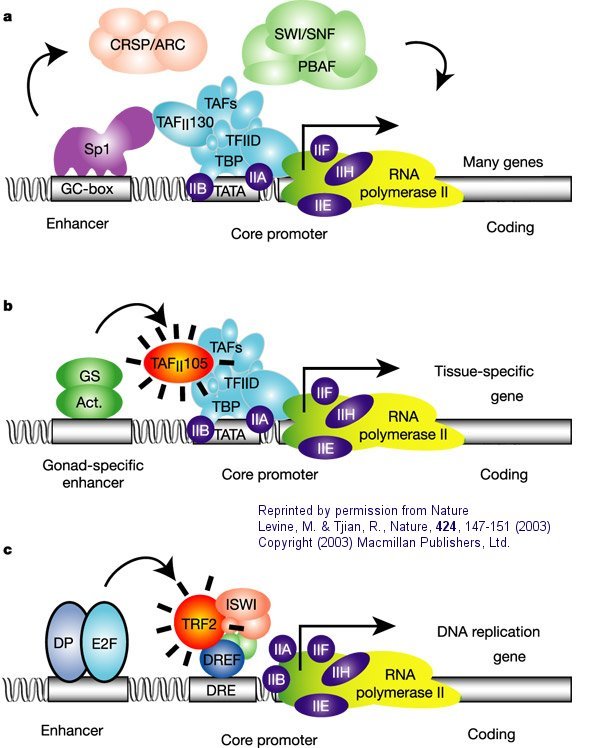
a. "The eukaryotic transcriptional apparatus can be subdivided into three broad classes of multi-subunit ensembles that include the RNA polymerase II core complex and associated general transcription factors (TFIIA, -B,-D,-E,-F and -H), multi-subunit cofactors (mediator, CRSP, TRAP, ARC/DRIP, and so on) and various chromatin modifying or remodeling complexes (SWI/SNF, PBAF, ACF, NURF and RSF)."
b, c. "Metazoan organisms have evolved multiple gene-selective and tissue-specific TFIID-like assemblies by using alternative TAFs (TBP-[TATA Binding Protein associated factors] such as the ovarian-specific TAF105) as well as TRFs (TBP-[TATA Binding Protein associated factors] related factors such as TRF2 in Drosophila and mice) to mediate the formation of specialized RNA polymerase initiation complexes that direct the transcription of tissue-specific and gene-selective programmes of expression." (Nature reference in figure above.)"
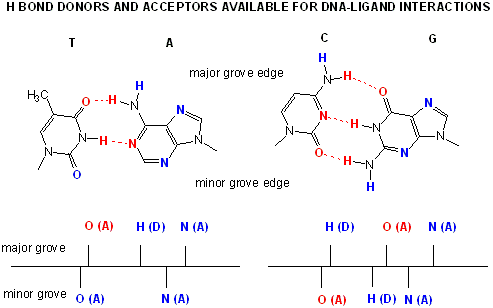
Proteins can interact specifically with DNA through electrostatic, H-bond, and hydrophobic interactions. AT and GC base pairs have available H bond donors and acceptors which are exposed in the major and minor grove of the ds DNA helix, allowing specific protein-DNA interactions.
Figure: AT and GC base pairs have available H bond donors and acceptors
![]() Jmol:
Updated Simple DNA Tutorial
Jmol14 (Java) |
JSMol (HTML5) (see last
selection buttons to see H bond donors and acceptors in the major grove.
Jmol:
Updated Simple DNA Tutorial
Jmol14 (Java) |
JSMol (HTML5) (see last
selection buttons to see H bond donors and acceptors in the major grove.
![]() Jmol:
Updated RNA Polymerase II/DNA/RNA
complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated RNA Polymerase II/DNA/RNA
complex
Jmol14 (Java) |
JSMol (HTML5)
Gene Transcription, Proteolysis and Membrane Lipids
An interesting example illustrates how transcriptional control maintains the balance of lipids in biological membranes. The phospholipids and sphingolipids in membranes are extremely heterogeneous, owing to the diversity of head groups and acyl chain composition. Given this great diversity, it is remarkable the different cells are able to maintain the specificity of lipid types in different cells, in different membranes within cells, and within a given leaflet of a membrane (remember our discussion of lipid rafts). How can the cell regulate the type of lipids that it synthesizes? What controls the transcription of genes for lipid synthesis?
Regulation of transcription of these genes appears to be controlled by multidomain proteins that bind to sterol response elements in the DNA. The proteins, called Sterol Response Element Binding Proteins (SREBPs) are activated by proteolysis to release a transcription factor domain which migrates to the nucleus. Proteolysis of SREBP occurs in the Golgi by resident proteases. The SREBP in the Golgi is in complex with another protein, SREP cleavage-activating protein (SCAP), which facilitates movement of the SREBP to the Golgi from its site of synthesis in the endoplasmic reticulum. Lipid regulation occurs when fatty acids, cholesterol, or PL derivatives like phosphoethanolamine (from ceramide) inhibits proteolytic activation of the SREBP. Regulation depends on whether or not SCAP "ferries" SREBP to the Golgi. SCAP binds to SREP and transfers it to the Golgi membrane, but only when sterol levels are low. When cholesterol is high, it binds to the transmembrane domain of SCAP and prevents SCAP from interacting with SREP and transferring it to the Golgi.
Auxin, a major plant hormone that induces gene expression, also seems to activate transcription through proteolysis. When bound to its soluble cytoplasmic receptors, ubiquitin protein ligase SCFTIR1, it activate proteolysis of protein that inhibit transcription.
![]() Jmol: Updated
Auxin Receptor
Jmol14 (Java) |
JSMol (HTML5) Auxin Receptor
Jmol: Updated
Auxin Receptor
Jmol14 (Java) |
JSMol (HTML5) Auxin Receptor
D5. DNA Binding Proteins
Given the relative structural simplicity and repetitiveness of DNA, it would follow that proteins that bind specifically to it might have common DNA binding domain motifs but with specific amino acids side chains allowing for specific binding interactions.
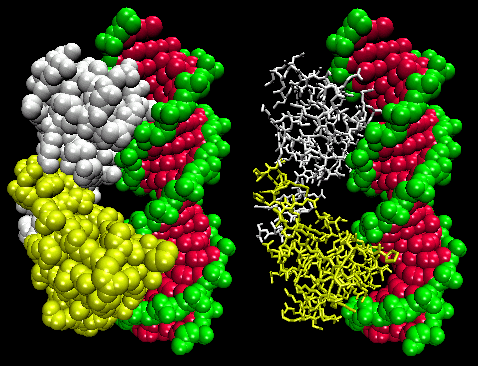
- helix-turn-helix: found in prokaryotic DNA binding proteins.
Figure: helix-turn-helix
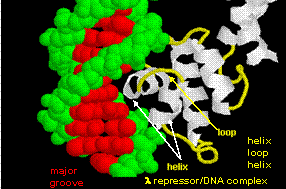
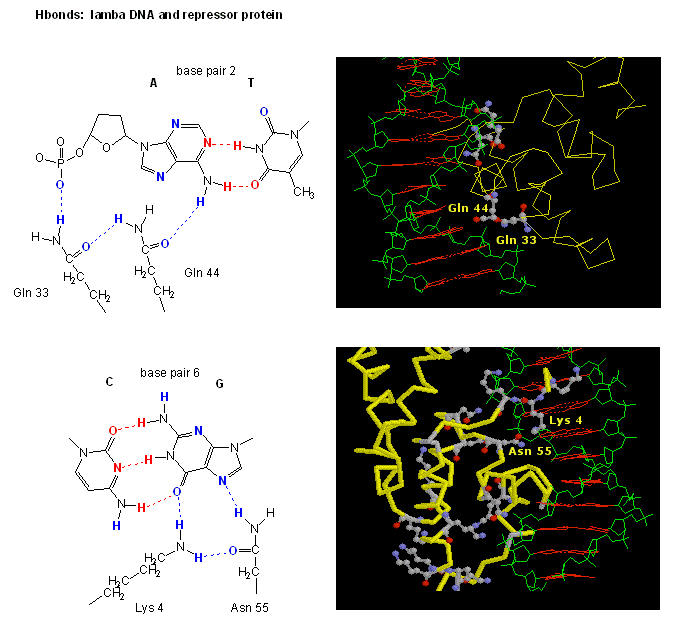
The figures shows two such proteins, the cro repressor from bacteriophage 434 and the lambda repressor from the bacteriophage lambda. (Bacteriophages are viruses that infect bacteia.) Notice how specificity is achieved, in part, by the formation of specific H-bonds between the protein and the major grove of the operator DNA.
Figure: Lambda Repressor/DNA Complex
Figure: H Bond interactions betweenλ repressor and DNA
{kind=link}
{kind=link}
![]() Jmol:
Updated Lambda Repressor/DNA complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Lambda Repressor/DNA complex
Jmol14 (Java) |
JSMol (HTML5)
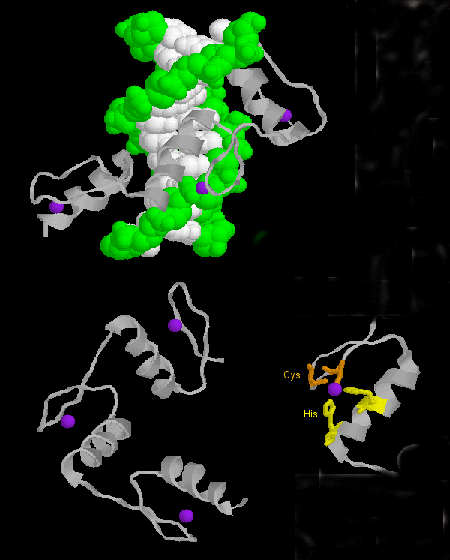
- zinc finger: (eukaryotes) These proteins have a common sequence
motif of
X3-Cys-X2-4-Cys-X12-His-X3-4-His-X4- in which X is any amino acid. Zn2+ is tetrahedrally coordinated with the Cys and His side chains, which are on one of two antiparallel beta strands, and an alpha helix, respectively. The zinc finger, stabilized by the zinc, binds to the major groove of DNA. ]
Figure: zinc finger
![]() Jmol:
Updated Zif268:DNA Complex
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Zif268:DNA Complex
Jmol14 (Java) |
JSMol (HTML5)
Zn finger proteins, of which 900 are encoded in the human genome (including the eukaryotic insulators binding protein CTCF described above) can be mobilized to actual repair specific mutations in cells, which if carried out in a high enough percentage of mutant cells could cure specific genetic diseases such as some forms of severe combined immunodeficiency disease. In this new technique (Urnov et al, 2005), multiple linked Zn finger binding domains, (one of the natural-occurring ones or mutant forms produced in the lab), each one specific for a certain nucleotide sequence, is linked to a nonspeciifc endonuclease, derived from the enzyme FokI. The nuclease is active in dimeric form so the active complex requires two endonuclease domains, each bound to four different Zn finger domains, to assemble at the target site. Specificity of binding is achieved by selection by the Zn finger domains. A nick is then made by the DNA by the nuclease, and host cell repair mechanisms ensue. This process involves strand separation, homologous recombination of the nicked region with complementary DNA within the cell, and repair of the nick. If excess wild type (non-mutated) DNA is added to the cells and uses as the template, the normal DNA repair mutation would fix the mutation. Urnov et al have shown the up to 20% of cultured cells containing a mutation can be repair in the lab. If these cells gain a selective growth advantage, the mutated cells would eventually be replaced with wild type cells.
- steroid hormone receptors: (eukaryotes) In contrast to most hormones, which bind to cell surface receptors, steroid hormones (derivatives of cholesterol) pass through the cell membrane and bind to cytoplasmic receptors through a hormone binding domain. This changes the shape of the receptor which then binds to a specific site on the DNA (hormone response element) though a DNA binding domain. In a structure analogous to the zinc finger, Zn 2+ is tetrahedrally coordinated to 4 Cys, in a globular-like structure which binds as a dimer to two identical, but reversed sequences of DNA (palindrome) within the major grove. (Examples of palindromes: Able was I ere I saw Elba; Dennis and Edna dine, said I, as Enid and Edna sinned.
Consider the glucocorticoid receptor (GR) as a specific example. It binds DNA as a dimer. The two DNA binding domains of the dimer associate with two adjacent major grooves of the DNA in the GR binding sequence (GBS), a short sequence of DNA within the promoter. Meijsing, et al. have found that not only does the GBS act as a binding site for GR, allowing transcription of genes, but it also affects the conformation of the receptor, causing gene transcription to be regulated in another way. The group constructed luciferase "reporters genes" which have GBS linked to the gene for the protein luciferase, that would express the protein luciferase (which fluoresces) if they were being transcribed, with the GBS. They found that relative transcriptional activity did not correlate to relative binding affinity of GR to the GBS. GBSs which were much more active than others bound comparably with those of lower activity, while GBSs with similar transcriptional activity bound with different affinities. This shows that the GBS is conferring unique function to the GR associated with it (i.e. transcription is not simply affected by whether or not the GR is bound to the GBS). A �lever arm� of the receptor was found to undergo conformational changes when bound to DNA, with changes specific to the sequence to which it was bound. A mutant protein, GR-γ, was made to be identical to the wild-type protein, GR-α, except in the lever arm was found to have different transcriptional activity even though they were binding to the same site on the DNA, showing that the lever arm and its conformation affects transcription.
- leucine zippers (or scissors): (eukaryotes) These proteins contain stretches of 35 amino acids in which Leu is found repeatedly at 7 amino acid intervals. These regions of the protein form amphiphilic helices, with Leu on one face, one Leu after two turns of a helix. Two of these proteins can form a dimer, stabilized by the binding of these nonpolar, leucine-rich amphiphilic helices to one another, forming a coiled-coil, much as in the muscle protein myosin. The leucine zipper represents the protein binding domain of the protein. The DNA binding domain is found in the first 30 N-terminal amino acids, which are basic and form an alpha helix when the protein binds to DNA. The leucine zipper then functions to bring two DNA binding proteins together, allowing the N-terminal bases helices to interact with the major grove of DNA in a base-specific fashion. Valine and isoleucine, along with leucine, are often found in stretches of amino acids that can interact to form other types of coiled coils.
Figure: leucine zippers (made with VMD)
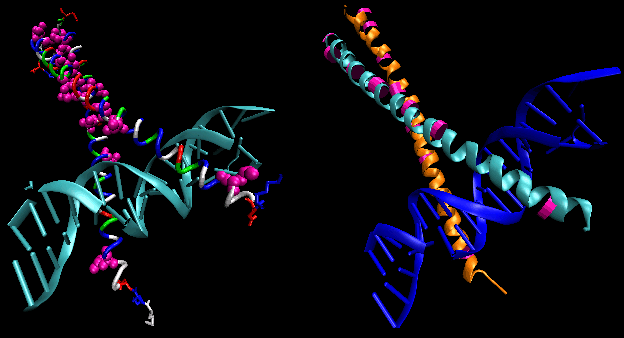
![]() Jmol: Updated
Leucine Zipper
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
Leucine Zipper
Jmol14 (Java) |
JSMol (HTML5)
Just as Zinc fingers nucleases have been used to induce repair of mutations, another study of the rat genome used specially designed ZFNs to cause breaks in ds-DNA that contain mutations from inaccurate DNA repair mechanism (by NHEJ) and hence contained specific mutations (Geurts, et al. 2009). This process, �knockout of the gene,� prevents the production of the protein normally transcribed by the target gene. Five- and six-finger ZFNs were used to achieve a high level of specificity in the targeted binding to the gene for three different proteins: green fluorescent protein (GFP), Immunoglobulin M (IgM) and Rab38. The knockout was successful in 12% of the rats tested; these animals had no wild-type protein and no expression. The ZFNs were sufficiently specific that no mutations were observed at any of 20 predicted non-target sites. This study supports the viability of control of transcription and expression for the treatment of disease and the importance of specific binding.
We have seen that two main factors contribute to the specific recognition of DNA by proteins; the formation of hydrogen bonds to specific nucleotide donors and acceptors in the major groove, and sequence-dependent deformations of the DNA helix to altered shapes with increased affinity of protein ligands. For example the Tata Binding Protein (TBP) can interact with a widened minor grove in the TATA box. New findings support that in addition proteins are able to use information in minor grooves that have become "narrowed" depending on the nucleotide sequence.
Tracks of DNA enriched in A can lead to twisting conformations that cause inter-base-pair hydrogen bonding in the major grooves, results in the narrowing of minor grooves. High amounts of AT base pairs are concentrated in narrow minor grooves (width <5.0 �) and CG base pairs are found more frequently in wide minor grooves.
How does minor groove narrowing affect DNA recognition? Narrow minor groves enhance the negative electrostatic potential of the DNA, making it a more specific and recognizable site. The backbone phosphates of the DNA are closer to the middle of the groove when it is narrow, thus correlating narrow minor grooves with a more negative electrostatic potential.
The minor grove-interacting parts of proteins contain arginine whose side chain can be accommodated into the more narrow and negative minor groove. Arginines can bind and in some cases insert themselves as short sequence motifs which enhance the specificity of the DNA shape recognition. Arg is preferred over Lys since the effective radii of the charge in Arg is greater than of the charge carrier in Lys. This would lead to a decreased desolvation energy for Arg which would promote its binding to the narrowed major grove. This discovery shows that "the role of DNA shape must be taken into consideration when annotating the entire genome and predicting transcription-factor-binding sites".
Figure: Arg in T3c Transposase binding in Narrowed Minor Grove of T3c Transposon
{kind=link}
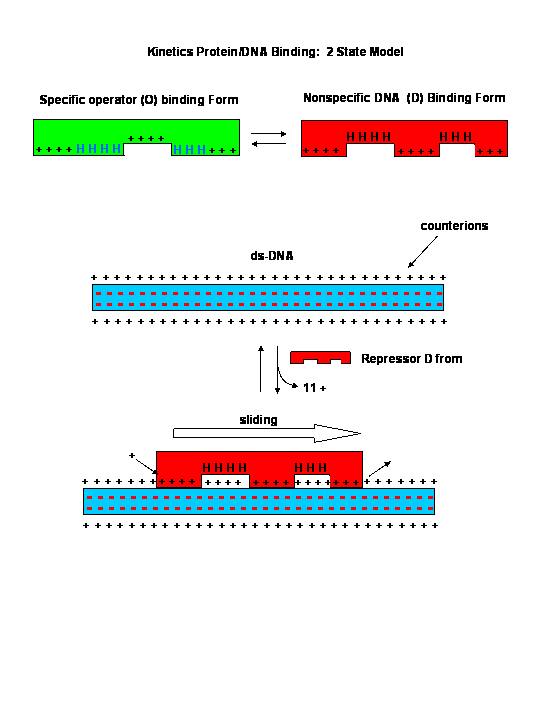
![]()
Sliding Model for protein/DNA Interactions
{kind=link}
D6. Studying the Interactome
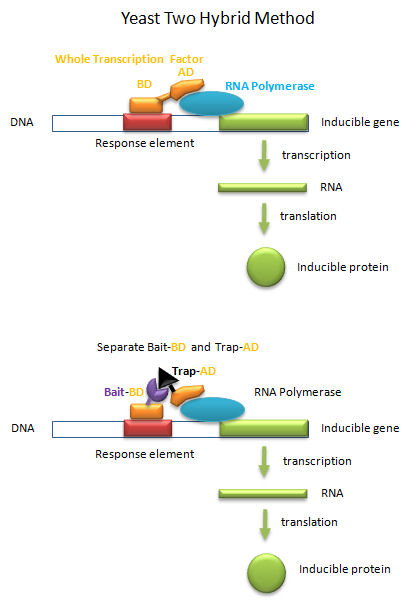
Yeast Two Hybrid System
Transcription factors must do more than bind to upstream targets (response elements) on DNA. They must also interact with and activate the transcription machinery, including RNA polymerase and other assembled proteins, for transcription to occur. To accomplish both tasks, protein transcription factors usually consist of two distinct and often separable domains, a DNA Binding Domain (DNA-BD) and an Activation Domain (AD). The BD usually contained the DNA binding motifs discussed above. In a clever feat of genetic manipulation, scientist have taken the part of the transcription factor gene encoding the DNA-BD and fused it to a gene for a protein called the bait protein. Likewise the other part of the transcription factor gene encoding the AD is fused it to a gene for another protein, the target, that could bind the bait protein. Plasmids with the gene constructs are added to yeast. By themselves, the separated BD and AD can not activate transcription from a gene in the yeast that is inducible by the whole transcription factor. However, if the BD-Bait gene and the AD-Target genes are both added on separate plasmids, and both fusion genes are ultimately transcribed and the fusion RNA translated into fusions proteins, then transcription from the inducible gene can occur if the bait and target part domains of the fusion proteins bind to each other, allowing binding of the BD domain and the AD domain to their target sites, leading to gene transcription. This yeast two-hybrid method has allowed the determination of protein binding partners, part of what is now termed the interactome.
Figure: Yeast Two-Hybrid
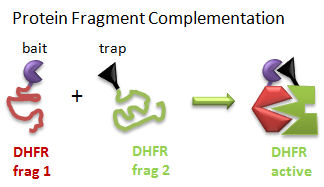
Protein Fragment Complementation (PFC) (Tarassov, K. et al): A gene encoding one fragment of a reporter enzyme, dihydrofolate reductase (DHFR), is fused to a gene for a bait protein and inserted into a plasmid. A second gene representing the second fragment of a reporter enzyme is linked to possible target protein genes. In a cell transformed with both plasmids, the reporter gene will ultimately display enzymatic activity only if the translated bait protein binds to a translated target protein, allowing the two fragments of the enzyme to interact and fold collectively into a holo-, active enzyme activity. The reporter gene was a mutant of DHFR that was resistant to an inhibitor, methotrexate. Functional DHFR activity leads to cell growth in the presence of methotrexate.
Figure: Protein Fragment Complementation
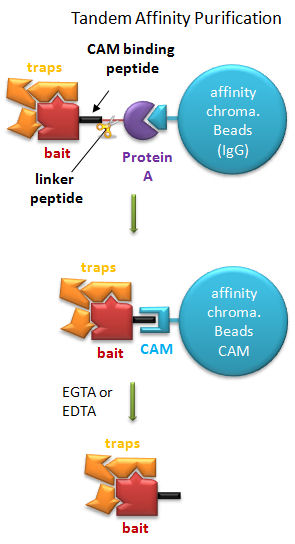
Tandem Affinity Purification (TAP) (Rigaut, G. et al) A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology 17. 1030 (1999): A gene for a bait protein is linked sequentially to genes encoding two separate tags, one for Protein A (which binds immunoglobulin G - IgG) and a calmodulin binding peptide (which binds the protein calmodulin in a process which requires calcium. In between the genes for the two tags is a nucleotide sequence encoding a short, protease sensitive linker peptide. The gene construct is introduced into yeast and expression induced. Cell are lysed and the extract applied to affinity chromatography beads containing covalently attached IgG, which binds the Protein A tag. After extensive washing the bound bait protein, with associated target proteins, is eluted by proteolysis of the peptide linker. The eluate is applied to a second affinity column containing covalently attached calmodulin. After washing, the trap and associated target proteins are eluted with a calcium chelator (EGTA or EDTA) as the interaction of calmodulin with the second tag, calmodulin binding peptide, requires calcium. Eluted target proteins can be identified by 2D PAGE or mass spectrometry.
Figure: Tandem Affinity Purification
A recent comparison of the Y2H, PFC, and TAP methods was made (Jensen, L. and Bork, 2008; Yu, H et al, 2008). Not unexpectedly, the Y2H was best at determining nuclear protein interactions, the TAP for cytoplasmic and abundant proteins, and PFC for transmembrane proteins. All suffer somewhat in identifying transient protein complexes.
D7. Phosphorylation and Control of Gene Expression
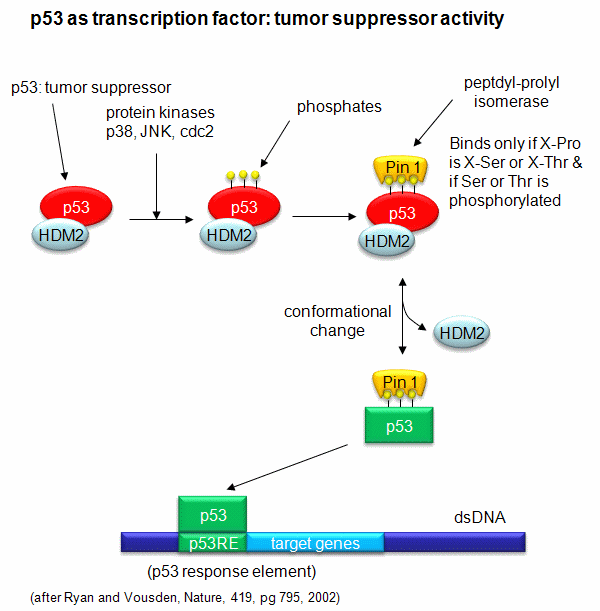
A common way to control gene expression is by controlling the post-translational phosphorylation of transcription factors by ATP. This modification might activate or inhibit the transcription factor in turning on gene expression. The added phosphate groups might be necessary for direct binding interactions leading to gene transcription or they might lead to a conformational change in the transcription factor, which could activate or inhibit gene transcription. A recent example of this later case is the control of the activity of the transcription factor p53. p53 has many activities in the cell, a primary one as a suppressor of tumor cell growth. It activates and inhibits a large number of genes in the process. If a cell is subjected to stress that results in genetic damage (an event which could lead a cell to transform into a tumor cell), this protein becomes an active transcription factor, leading to the expression of many genes, including those involved in programmed cell death and cell cycle regulation. Both of these effects could clearly inhibit cell proliferation. Hence p53 is a tumor suppressor gene. p53 is usually bound to the protein HDM2 which down regulates its activity by leading to its degradation. Stress signals lead to the activation of protein kinases in the cell (such as p38, JNK, and cdc2), causing phosphorylation of Ser 33 and 315 and Thr 81 in p53. This leads to the binding of Pin 1, a peptidyl-prolyl isomerase, which catalyzes the trans<=>cis conformational changes of X-Pro bounds. Pin 1 appears to bind only when p53 is phosphorylated. The ensuing change in p53 conformation presumably leads to its activation as a transcription factor.
Figure: Activation of p53 as a transcription factor by phosphorylation and conformational change
p53 binds to the p53 response element (p53RE) as shown above. In doing so it could either activate or repress gene transcription. The response element consists of two, 10-mers separated by a spacer of 0-13 nucleotides, and has the following canonical structure: RRRCWWGYYYspacerRRRCWWGYYY with W = A or T, R = C or G, and Y = C or T. The CWWG motif is required for high affinity binding of p53 to the response element. Alternations in the other nucleotides would fine tune binding and probably direct p53 to activate or repress transcription from the promoter. Over 1500 genes with a p53RE have been identified with many of these being repressed on binding of p53. Over 500 different high affinity binding sites for p53 have been found using microarrays. Wang et al have systematically altered the nucleotides in the p53RE linked to a promoter controlling the luciferase gene (which on activation and production of the protein luciferase can be monitored by light emission on addition of the substrate lucifein which releases a photon on reaction with dioxygen. They chose p53RE and promoters for two different genes, p21 whose transcription is known to be activated by p53, and Lasp1whose transcription is repressed. A figure representing the canonical sequence for both activating and repressing p53RE is shown below. (See the legend below for detailed explanation.) The dinucleotide sequence between the canonical C and G in each 10mer is a major determinant of whether p53 leads to activation or inhibition for transcription from promoter controlled by the response element.
Figure: Canonical sequence for both activating and repressing p53RE
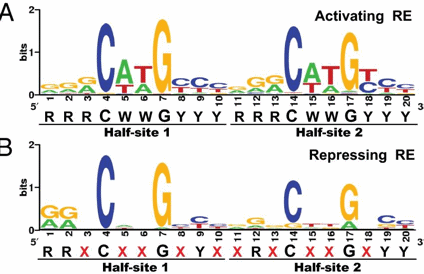
From Wang, Bei et al. Redefining the p53 response element. PNAS 2009 106 (34) 14373-14378; doi:10.1073/pnas.0903284106. Permission: Anyone may, without requesting permission, use original figures or tables published in PNAS for noncommercial and educational use (i.e., in a review article, in a book that is not for sale) provided that the original source and the applicable copyright notice are cited,
Each weblogo above (http://weblogo.berkeley.edu/logo.cgi ) consists of stacks of letters, one stack for each position in the sequence. The overall height of each stack indicates the sequence conservation at that position (measured in bits), whereas the height of symbols within the stack reflects the relative frequency of the corresponding amino or nucleic acid at that position.
The repressive capabilities of the p53 protein have been studied quite extensively, yet little work seems to have been done surrounding what, if any, secondary mechanisms may affect the way it functions. The work of Wang et al attempts to address this problem. Through various substitutions of the base pair sequence they uncovered that two main elements seem to affect the function of this protein. First, the arrangement of a dinucleotide promoter directly affects whether or not p53 acts as a repressor or an activator. Some combinations will guarantee a certain function, whereas others depend more of the half-site background. Overall, the dinucleotide core seems to play an important role in the stabilization of the p53 to its response element. The second main way the function of the p53 can be modified is by the lateral triplet flank motifs. This implies that certain triplets of nucleotides located specifically in the base pair sequence can code for great activation. Determined by substitution of a �comprehensive set of synthetic triplet flanking motifs�, the RRR-YYY:RRR-YYY combination seemed to give a maximal activation. Moreover, they authors reviewed functions of previously reported p53 proteins. They found that some proteins thought to be activating were actually repressing, and some that were thought to be repressing, were actually activating. As a result, they insisted on paying greater attention to the base pair sequence, and avoiding deviations. After examining their study, it is clear that the p53 protein cannot continue to be defined as a single function protein. Rather, based on the evidence presented by the authors, the protein can act as either a repressor or an activator. This change of focus implies that it is necessary to reassess the previously analyzed p53 proteins to examine the true nature of their function.
D8. Classification of Transcription Factors
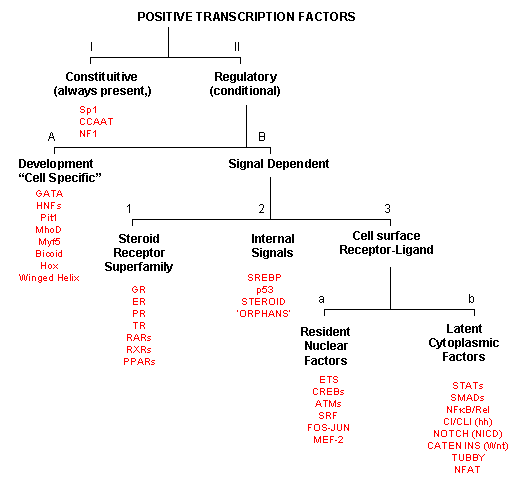
As inferred from above, transcription factors can be classified based on their protein structure. A newer classification scheme, based on the function/activity of the transcription factors, has been proposed by Brivan, Lou and Darnell (Science, 295, pg 813, 2002), as illustrated in the flowchart below, along with specific examples. The classes of transcription factors include those that are:
-
constitutively active : are always active in the nucleus of the cell and probably activate transcription of genes that must always be turned on;
The rest must be activated by some means, which include those that are:
-
developmental or cell type-specific whose genes must be transcribed (probably in a regulated fashion) to form the transcription factor which then enters the nucleus;
-
signal dependent transcription factors, which are activated through a signaling event.
There are classes of signal-dependent transcription factors that are activated by:
-
steroids, which are cholesterol derivatives that can pass through the cell membrane and bind steroid-specific transcription factors which turn on specific sets of genes; most of these transcription factors are present in the nucleus and are activated there by steroid hormones. One exception is the glucocorticoid receptor (GR) which is found in the cytoplasm;
-
internal signals derived from the cell, such as internally made lipid signals.
-
cell surface receptor-ligand interactions;
There are two types of receptor-ligand interactions that lead to transcription factor initiation.
-
small ligand molecules (like epinephrine) bind transmembrane receptors which leads to formation of second messengers or signals inside the cell, which ultimately activate Ser-phosphorylation activity. Nuclear transcription factors can become phosphorylated and activated.
-
small ligands bind transmembrane receptors which then bind to and activate latent transcription factors in the cytoplasm, which then migrate to the nucleus.
Figure: Transcription Factors: Functional Classification
D9. Cooperative Binding of Proteins to DNA
We have just spend much time studying the cooperative binding of oxygen to hemoglobin. Cooperativity seemed to be require conformational changes in a multimeric protein. Is it possible to get cooperative binding of ligands without conformational changes? In a recent book by Ptashne and Gann (Genes and Signals, Cold Spring Harbor Press, 2002), it is argued that you can and through a very simple mechanism.
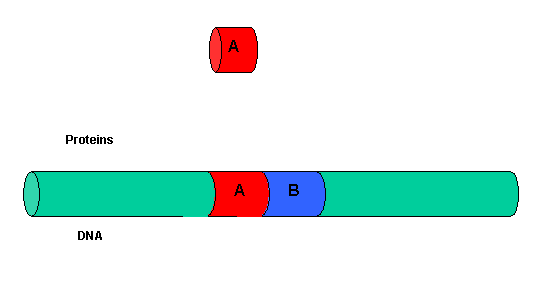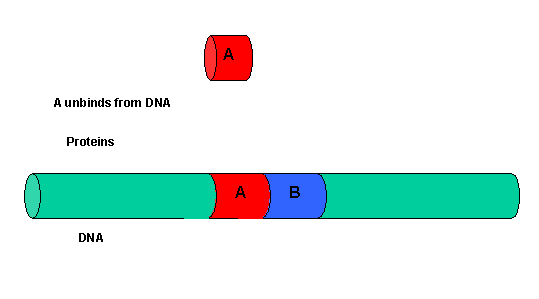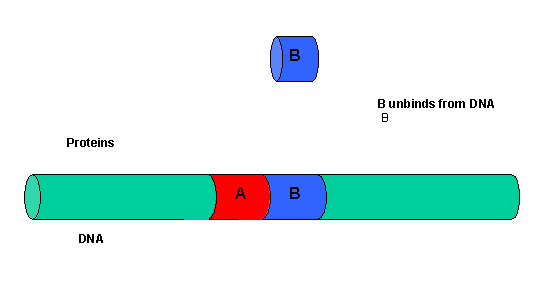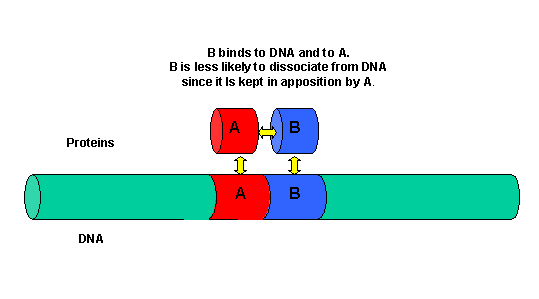
It must be clear that to activate gene transcription, several transcription factor proteins must assembly at the promoter before RNA polymerase can transcribe a gene. There are multiple DNA-protein and protein-protein contacts. To simplify this discussion, consider the case of two proteins, A and B, that must bind to the DNA and to each other for transcription to occur.
Figure: two proteins, A and B

The binding of each protein alone is characterized by a characteristic Kd, kon, and koff. What happens to kon and koff for protein B, for example, when A is already bound? You can imagine that kon doesn't change much, but what about koff after the protein is interacting both with its DNA site and with protein A? If B did dissociate from its DNA site, it would still be held in close approximation to that site because of its interaction with the bound protein A. Its effective concentration goes up and you should readily image that it would rebind very quickly to its DNA site. The net effect would be that it's apparent koff would decrease, which would increase its apparent binding affinity and decrease its apparent Kd (remember that Kd = koff/kon). Hence prior binding of A would lead to cooperative binding of protein B.
- Animation showing cooperative binding of B to the protein A-DNA complex.
-
 Nuclear
receptor Database
Nuclear
receptor Database
{kind=link}
D10. Epigenetic Control of DNA - Methylation
The fertilized egg is a totipotent cell. That is, through a series of divisions, its progeny cells can eventually become any of about 200 histologically different cell types. With subsequent cell division in the developing embryo, cells find themselves in different topological environments and have different cell-cell contact. Through signal transduction through the cell membrane, these cells start to become different, or differentiate, into other cells types. They do so by activating and inhibiting the expression of a different set of genes to form a different set of proteins in the cells. Most cells become terminally differentiated and eventually (after maybe a hundred cell divisions) lose the ability to divide and hence begin to die. However, a few types of cells, called stem cells, retain the ability to differentiate into other cells types in a regenerative process. These cells are pluripotent in that they can differentiate into other cell types.
How do dividing cells know what types of genes to actively transcribe? How can they have "memory" of the cells type they were before division? This appears to happen without alteration of the nucleotide sequence of the DNA in these cells. The main mechanism appears to be a inheritable but modifiable pattern of chemical modifications to the DNA (not unlike co- or post-translational modification of proteins) involving methylation/demethylation (by a methylase and demethylase) of cytosine in CpG dinucleotide repeats in the DNA. Also proteins can bind to DNA and methylated DNA to modify the course of gene expression in daughter cells. Such chemical modifications to the DNA which modify gene transcription are examples of epigenetic changes in the DNA.
You are all familiar with the cloning of animals from the DNA of adult cells (from Jurassic Park to the actual cloning of the sheep Dolly). In this process, the nucleus from an adult somatic cell (like a check epithelial cell) is removed and placed in an egg from which the nucleus has been removed (enucleated). The egg now has a full complement of DNA, just like an adult cell, but it didn't get the full set by normal means - i.e. by receiving half from a sperm to complement its own normal half. The DNA in the egg also has the methylation pattern of a terminally differentiated adult cell. It must be reprogrammed by undergoing extensive demethylation and remethylation to the "correct" epigenetic methylation state if the egg has a chance to form a normal embryo, fetus, and adult. Obviously this can happen, as evidenced by Dolly and success in cloning cows, cats, pigs, mice, and dogs. However, it is very difficult to achieve and probably accounts for the low success rate of cloning.
Methylation patterns can account for gene silencing (in which one gene in a pair of identical chromosomes is not expressed) and inactivation of one entire X chromosome in a female (who has 2 X chromosomes). In general, transcription from genes that are methylated is inhibited.
![]() Jmol: Updated
Methyl-CpG-Binding Domain of Human MBD1 in Complex with Methylated
DNA
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
Methyl-CpG-Binding Domain of Human MBD1 in Complex with Methylated
DNA
Jmol14 (Java) |
JSMol (HTML5)
Methylation patterns are inheritable and are also determined by the environmental variables. A recent study of neuron specific glucocorticoid receptor gene (NRGC1) promoter in the hippocampus showed an increase in methylation of the promoter and decreased levels of the mRNA transcript from the gene in suicide victims who had a history of childhood abuse compared to suicide victims that didn't suffer such abuse. These result paralleled those found in rats who were raised in a non-nurturing environment. The hippocampus is involved in the stress response.
Methylation patterns can be passed onto to daughter cells and on to offspring. This inheritance depends on specific binding of the DNA methylase to the methylated CpG on the template strand of the newly replicated DNA, which positions it to methylate the complementary newly replicated strand on the C hydrogen bonded to the template strand G. This enzyme specificity explains the requirement for a CpG dinucleotide methylation site in DNA, as shown below.
Figure: Methylation of CpG
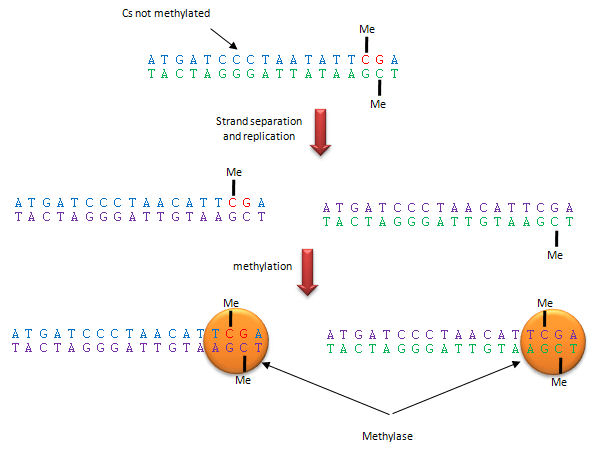
The dinucleotide sequence CpG is underrepresented in the human genome, yet is found in about 60% of promoter regions for genes. CpGs in promoter regions that are constitutively transcribed are not methylated. Methylated DNA outside of genes for proteins help to silence transcription from those regions. The CpG dinucleotide is probably found in low abundance since if 5 methyl cytosine spontaneously deaminates, it forms thymine which would lead from a CG to AT base pair mutation.
Yet another covalent epigentic modification has been discovered in neurons, where high levels of 5-hydroxymethyl cytosine are found. A 5-methylcytosine hydroxylase has also been discovered. Both probably have large roles in gene regulation.
D11. Chromatin Remodeling and Gene Expression
Control of DNA transcription in eukaryotes was thought to involve the assembly of many proteins at the promoter into a pre-initiation complex (PIC). Once assembled, RNA polymerase could bind and transcription would be initiated. But wait a minute! Isn't DNA packaged in the nucleus into chromatin in which 147 BP of DNA is wound around a core of 4 pairs of positively charged histone proteins - including H2A, 2B, 3, and 4 - to form a nucleosome, seen under a microscope as beads on a string?
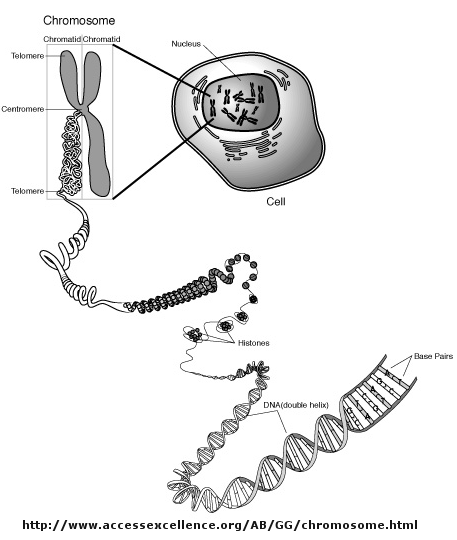{kind=link}
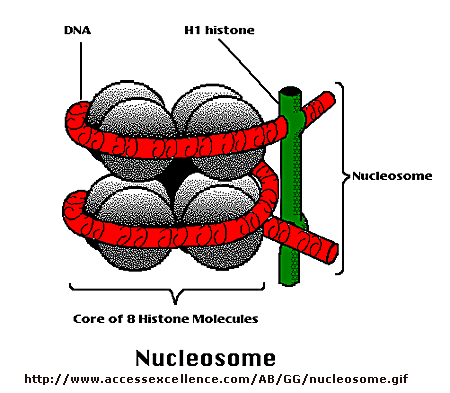{kind=link}
![]() Jmol:
Updated Nucleosome
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Nucleosome
Jmol14 (Java) |
JSMol (HTML5)
Isn't this chromatin further wound into fibers which result in the classic picture of sister chromatids ready to separate at cell division?How could the transcription factors and RNA polymerase recognize target sites on DNA given this degree of "folding" and condensation of the DNA?
-
Animation:
Packing of DNA from the Walter + Eliza Hall Institute of Medical
Research
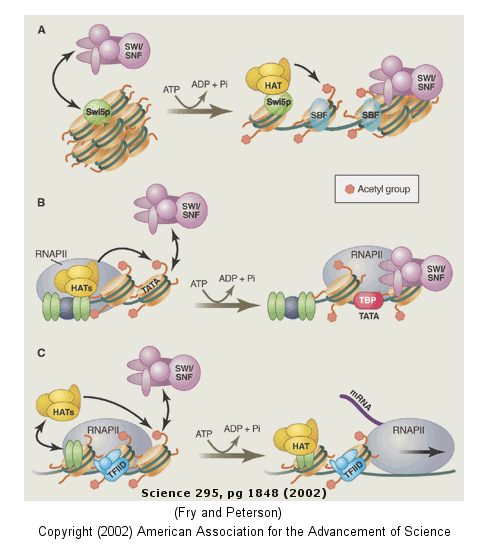
Clearly the complex compacted state of DNA and its interaction with the histone proteins must be "remodeled" to allow interactions of the transcription factors and RNA polymerase (which is about the same size as a nucleosome). The regulation of this chromatin remodeling clearly affects gene transcription, and is another example of epigenetic changes that can affects phenotype. The state of chromatin structure is regulated by enzymes that affect histone structure and function by chemically modifying the histone proteins (through acetylation, methylation, and phosphorylation) . Likewise, the DNA at the promoter region is changed by enzymes that remodel the DNA through an ATP dependent series of modifications. For example when histones are modified by histone acetyltransferase (HAT's), other modeling factors (SWI/SNF) are recruited to the chromatin. Chromatin remodeling would also be affected by that cell cycle stage of the cell. For example, chromatin condensed in sister chromatids ready for cells division would have different remodeling requirements for gene transcription than might chromatin in the form of bead on a string. Likewise remodeling efforts would also be gene-specific.
The figure below shows how remodeling is coupled to formation of the pre-initiation complex for three genes:
-
yeast HO gene: Swi5p activator binding results in the interaction of the SWI/SNF ATP-dependent remodeling enzyme, which leads to the binding of histone acetyltransferase (HAT). These facilitate formation of the pre-initiation complex.
-
human interferon-β gene: gene sequences known as activators, 5' to the promoter, bind HATs. When histones are acetylated, SWI/SNF interacts to remodel the chromatin and facilitate PIC formation.
-
human α-1 antitrypsin gene: the PIC is preformed and recruits HAT and SWI/SNF, which leads to gene transcription.
Alternations in chromatin remodeling could lead to changes in gene expression, in some cases causing cancer. SNF5 is a component of the SWI/SNF complex and in its normal form acts to suppress tumors (i.e. its gene is a tumor suppressor gene). Mutations in SNF5 are associated with rare and aggressive childhood tumors. Stuart Orkin has developed a technique to alter the gene in some mouse cells to produce an inverted gene which produces no functional SNF5. Cells with this mutation become tumor cells almost immediately.
Figure: Remodeling of Chromatin and Control of DNA Transcription
DNA winds around the histone core to form the nucleosome. However, histone tails not associated with DNA binding protrude from the nucleosome, and the function of these tails is just being unraveled. The amino acids in these tails are clearly sites for posttranslational modifications, including methylation, acetylation, and phosphorylation. When modified, these tails would provide additional binding sites for protein which could regulate transcription and chromatin modeling, thus modifying the "genetic code". Understanding the "histone code" and how it affects gene transcription becomes important. For example, the methylation of Lys 9 on histone 3 leads to binding of heterochromatin-associated protein, leading to inhibition of gene transcription (an example of epigenetic silencing). Acetylation of the tails generally leads to activation of gene transcription at that site. Acetylation of Lys residues converts them to amides and removes the positive charge of the amine. This would lead to decreased electrostatic interactions between the DNA and histones proteins, making the DNA more available for interaction with transcription factors and RNA polymerase.
Epigenetic changes (through methylation of DNA or acetylation, methylation, and phosphorylation of histone proteins) causing chromatin remodeling may change phenotype (characteristics of the individual) as evidenced by the fact that identical twins can eventually diverge in ways that effect their propensities to disease. Differences in diet and lifestyle, which can alter disease propensity, might exert their effects through epigenetic changes in gene expression. The Human Epigenome Consortium is developing a catalog of methylation pattern differences in the human genome which might be correlated with disease risk.
The nucleosome core is about the same size as RNA polymerase. How can RNA polymerase bind to its promoter site if it is wrapped around a nucleosome? One obvious answer is that nucleosome are not evenly distributed on chromosomal DNA, and perhaps not even found at promoter sites on the DNA. Rando et al. have studied the distribution of nucleosomes along the yeast genome. They cleaved internucleosomal DNA with nucleases leaving behind the nuclease protected-DNA. They separated the bound DNA from the nucleosome proteins, and labeled it with fluorescein. Next, total yeast DNA was isolated, fragmented, and labeled with rhodamine. They added both fluorescently labeled fragments to microarrays situated with overlapping 50 bp yeast chromosome 3 fragments. Equal red and green fluorescence at a given site on the array would arise if the DNA fragments labeled with fluorescein were protected by the nucleosome protein core particle. Low green to red fluorescence would arise if the fluorescein-labeled DNA was not protected by the nucleosome core.
From a thermodynamic viewpoint, binding affinities for the nucleosome protein core should be the same anywhere along the chromosomal DNA. This would lead to the prediction that nucleosomes would bind randomly along the DNA at all locations. leading to a constant ratio of green to red fluorescence across the array. That is, there would not be district signals from the array, but rather a smeared-out signal when the DNA was extracted from many yeast cells. The actual data showed sharp fluorescein/rhodamine signals and was consistent with fact that 70% of the nucleosomes were positioned at the same position in the DNA in different cells. Promoter sites for active genes were generally not occupied by nucleosomes. It was unclear if these sites are always free of nucleosomes or whether protein transcription factors and RNA polymerase cause the nucleosomal core proteins to slide away from the promoter sites.
Recent work suggests that positions of nucleosomes along the DNA is encoded in part by the DNA sequence itself, adding yet another "genetic code" that controls gene expressions. DNA must bend around the nucleosome core. Certain dsDNA sequences are more bendable that others, and the would be expected to have a greater chance of being involved in nucleosome complexes and less accessible for transcription. Segal et al isolated nucleosome bound DNA sequences and developed a computation model to predict which sequence of DNA would be bendable and hence be able to easily form nucleosome complexes. In other words, they calculated which DNA sequences would have high affinity for nucleosomes. They concluded that 50% of the positioning of nucleosomes can be accounted for by certain DNA sequences having higher affinity of the histone octamer. They found low nucleosome occupancy at important regulatory sites such as transcription initiation sites. Regions of the chromosome coding for tRNA and rRNA, which are highly expressed, were found to have low nucleosome occupancy.
D12. Acetylation and Methylation of Histones
Acetylation of histones is obviously an important method in the control
of gene transcription. A recent study by Choudhary et al investigated
the effect and prevalence of lysine acetylation in a range of other cellular
pathways. The study discovered over 3600 acetylation sites on 1750 different
proteins comprising the acetylome using high resolution mass spectrometery.
These regulate a wide variety of dissimilar cellular function and showed
acetylation as a prevalent form of post translational modification falling
in terms of its frequency between the phosphoproteome and the spectrum of
ubiquitinated proteins. It is a highly conserved process occurring in many
different cellular lines from prokaryotes to human beings, and being as
prevalent as phosphoproteins found in the evolutionary tree.
Acetylation eliminates the positive charge on lysine side chains in a
reversible process. It has already been established that acetylation of
lysine side chains was a key component of DNA damage repair as it modifies
histone protein tails found in the DNA. However, recently it has been shown
that acetylation�s effects extend to regulation of other cellular functions.
Most commonly acetylation plays a role in nearly all nuclear functions, but
it also plays a surprisingly big role in cytoplasmic functions. One of the
new cellular functions investigated was the involvement of acetylation in
regulating macromolecular complexes within the cell pertaining to functions
such as signal transduction, DNA damage repair, and the cell cycle. One
example protein included in the study is the 14-3-3 protein which binds
specifically to phosphoserine or phosphothreonine in phosphorylatedpeptides.
Four different lysines in the protein were mutated to glutamine in an
attempt to determine the effect of acetylation on the protein's binding.
Acetylation was found to regulate binding as the enzyme�s activity was
severely harmed by mutation. This has been seen in other cellular processes
where acetylation has lead to regulation of enzymatic activity. In
addition, the study discovered that there is important interaction between
phosphorylation and acetylation. This interaction or �cross talk� between
acetylation and other post translational modification methods in regulating
cellular activity has been observed in the protein p53 as well which plays
an important role in repairing damaged DNA.
-
- erasers: enzymes like histone deacetylases and lysine demethylases
- readers: proteins that recognize and bind to post-translationally modified histones including bromodomain-containing proteins (which recognize methylated Lys side chains), and methyl-Lys and methyl-Arg binding domain proteins
These protein are increasingly becoming targets for drug design as a way to alter gene transcription.
D13. Control of Gene Expression by RNA
What accounts for the increased complexity of organisms like humans? As was discussed in the DNA chapter, it is not the number of chromosomes or even the number of possible genes in an organism. One big difference between bacterial and human cells, for example, is the percentage of DNA coding for proteins. In bacteria, most of the DNA codes for proteins, but in human eukaryotic cells, most of the DNA (up to 98%) is "junk" in that it does not code for proteins. The DNA consists of intervening sequences within DNA coding for a given protein, and sequences between genes. Up to 98 % of the RNA transcribed in human cells is derived from this "junk" DNA. What function does this RNA serve? New evidence shows that this transcribed RNA binds to other RNA molecules like mRNA (to inhibit its translation), to DNA (to control gene transcription) or to proteins (to alter gene transcription as well). These process are called RNA interference (RNAi).
An understanding of RNAi really began in 1998 with the study of RNAi in round worms (described below) by Fire and Mellon, who were awarded the Nobel Prize in Physiology and Medicine in 2006. The Nobel Foundation stated that "the discovery of RNAi has already had an immense impact on biomedical research and will most likely lead to novel medical applications in the future".
The terminology used to describe the RNA species involved in RNAi is often confusing (especially to a protein chemist). In part it depends on the sources of the RNA. The RNA can derive from the cells own endogenous RNA (transcribed from genes of the cell) or by exogenous RNA entering the cell from the outside. This can happen by infection by an RNA virus, a DNA virus (which forms RNA in the cell after transcription), by plasmids containing DNA that will be transcribed to RNA in the cell, or by synthetic RNA. These RNAs generally inhibit translation of mRNA for a given protein and are described below in more detail.
RNAi from enodgenous RNA
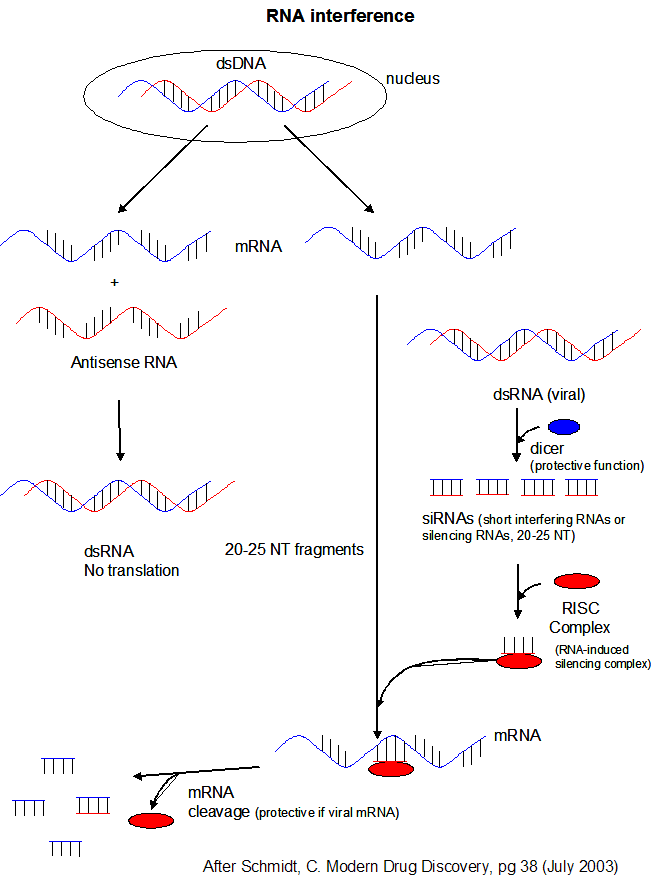
- microRNAs (miRNAs) are formed from endogenous RNA arising from transcription of certain stretches of nuclear DNA. From these regions, the transcribed RNA can base pair on itself to form a double stranded stem-loop hairpin structure. The initial transcript is call primary miRNA (or pri-miRNA). The pri-miRNA are transcribed by RNA polymerase III and are post-transcriptionally process to contain a modified 5' nucleotide and a 3' poly A tail The pri-miRNA folds to form regions stem-loop structures where the ds-stem is held together by intracchain H bonds. This stem-loop motif is recognized by an enzyme called Drosha, which cleaves it to form a separated stem:loop structure of about 70 nucleotide (NT) size (often called a pre-miRNA) with nonoverlapping ends. The stem:loop structures also can be called short hairpin RNA (shRNA). The dsRNA of the stem loop is recognized by an enzyme called dicer which removes the loop leaving a dsRNA of about 20-25 NTs in length. The short dsRNAs can then bind to a protein complex called RISC (RNA-induced silencing complex), which promotes unraveling of the dsRNA. A complex of RISC and one of the short RNA strands can bind to complementary stretches in mRNA for a specific gene (viral or host) and inhibit translation of the mRNA into protein. Inhibition occurs when RISC complex cleaves the RISC-siRNA- mRNA complex. (i.e one of the components in the RISC complex is an RNA nuclease.) This mechanism might be linked to defense mechanisms of virally-infected cells by inhibition of viral mRNA translation. If the complementarity of the mRNA and miRNA is less than ideal (which it usually is for miRNA), then binding of miRNA to the mRNA may only attenuate the translation of the mRNA. miRNA are often complementary to the 3' untranslated region (UTR) of the mRNA. It is estimated that the human genome has over 1000 miRNA which are used to regulate gene transcription (through attenuation of protein synthesis.
RNAi from exogenous RNA (or DNA)
- short interfering RNAs (siRNAs): These are synthesized in the lab and can be used to interfere with translation when added to cells. They are made to be perfectly complementary to a mRNA target. Often three different siRNA are used to target different regions of the target mRNA. As these are synthetic, their main function is in experimental studies to probe the effect of knocking down the expression of a gene by temporarily decreasing translation from a specific mRNA. These knockdowns are temporary as the siRNA degrade in the cell and are not replicated. Modified bases can be used in the synthesis of the siRNA to increase their half-life in the cell. It is difficult to get siRNAs into the cell (in a process called transfection) to produce these alterations in gene expression. Special transfection reagents have been developed to facilitate that process.
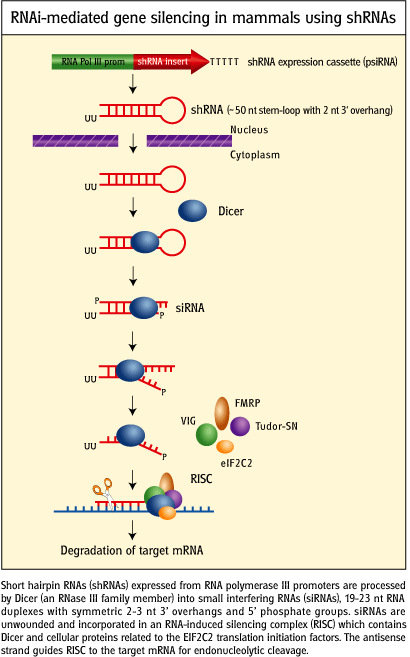
- short hairpin RNA (shRNA): instead of adding siRNA directly, a plasmid DNA containing a gene that when transcribed in the cell can form a shRNA can be added to the cell. Uptake of plasmids containing human genes by prokaryotes (in a process called transformation) is the most common way to make human proteins in bacterial cells. The plasmids can be replicated so the transformation of cells that take up the plasmid is passed on to progeny cells. Likewise eukaryotic cells can be transfected (unfortunately the word transformation is not used for eukaryotic cells) by plasmids with genes encoding shRNA. Knockdowns produced by this technique would be much longer lived that those produced by the addition of synthetic siRNAs.
- lentiviral transfection with shRNAs: Lentiviruses (like HIV) contain a ssRNA genome. It can be modified to contain a gene for a shRNA. Once in the cells, the lentilviral RNA can be converted to DNA which is integrated covalently into the host cell genome. When transcribed, shRNA is produced which when processed byh diceer produces RNA interference in the cell. This transfection of cells is essentially permanent, allowing knockdowns of protein expression in progeny cells.
RNAi pathways probably evolved from or with pathways for cellular resistance to viruses. Viruses often produced dsRNA during their life cycle. Some viruses like the HIV virus have a ssRNA genome. One method of host defense against viral infection is fomation of short interfering RNAs that could inhibit transcription of viral proteins from viral mRNA.
![]() Jmol:
dsRNA (still figuring out to convert to JSMol)
Jmol:
dsRNA (still figuring out to convert to JSMol)
One of the first amazing demonstrations of the power of RNAi to module gene expression at the translational level was done in the nematode worm, C. elegans. This organism has about 20,000 genes which code for proteins. Kamathk et. al. fed these worms E. Coli transformed with plasmid DNA designed to produced dsRNA upon transcription, one strand of which was complementary to mRNA sequences in the worm. Plasmids containing almost 17,000 different dsRNA encoding genes were constructed and used to knockout gene expression by forming dsRNA complexes of the mRNA with the RNAi. Phenotypic changes in the organism were studied. About 1700 of the dsRNA experiments led to observable (phenotypical) changes in the organism. Genes whose inactivation was lethal (and hence were essential for survival) were generally those that had counterparts in all other organism, while those associated with nonlethal changes were more likely to be homologous to genes in higher organisms and more recently evolved. They also selectively looked at which genes influenced lipid metabolism by incorporating a fluorescent tag which bound to lipid deposits in the organism. Around 300 genes were found to influence fluorescence and hence regulate fat deposition in the organism.
Figure: RNA Interferene: Antisense and Silencing
-
Animation:
RNAi Web
RNAi is the basis of an new emerging industry. Many companies offer kits and free software that make RNAi studies simple. Invitrogen is one such company.
Figure: RNAi-mediated gene silencing in mammals using short haripin
RNA genes.
credit:
http://www.invivogen.com/sscat.php?ID=14
![]() Jmol: Updated
Dicer
Jmol14 (Java) |
JSMol (HTML5) Dicer
Jmol: Updated
Dicer
Jmol14 (Java) |
JSMol (HTML5) Dicer
Two groups have deleted miRNA-155 and looked at effects on immune cells in mice. Immune cell function in B, T, and dendritic cells was affected, leading to animal death when exposed to salmonella after they were immunized. Animals in sterile environments showed no effect. In contrast to knockouts of protein-coding genes, these knockouts affect transcription of multiple genes. Knockout of miR-208 caused heart problems in mice placed in a stressful environment. These experiments indicated that some genetic diseases might arise from mutations in non-protein coding regions of the genome.
Recently, a new mechanism in control of gene expression has been offered which involves regulation of translation of a mRNA. mRNA must have a sequence, the Shine-Delgarno sequence, which allows it to bind to ribosomes. If a ligand binds to this site, mRNA could not bind to the ribosome and translation would be inhibited. Such is the case in the mRNA encoding proteins involved in the transport and synthesis of vitamins B1 (thiamine) and B12 (adenosyl cobalamin). Thiamin and thiamine pyrophosphate were shown to bind to the leader sequence of an E. Coli mRNA involved in thiamine biosynthesis and inhibit the translation of the mRNA. This allosteric mechanism for inhibition makes physiological sense since the presence of high levels of cellular B1 would obviate the need for its synthesis or transport.
D14. Recognition of viral and bacterial DNA by the immune system
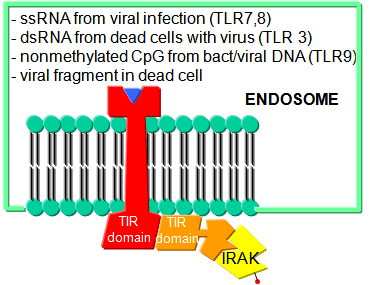
Before leaving the topic of RNA/proteins interactions, consider how a self cell would detect viruses and bacteria. It would be beneficial to the organism if the immune system could recognize and response to many types of bacteria, viruses, fungi, or protozoa by binding to common target on them. For example, it would be desirable to have a single cell type, such as a scavenging macrophage, have a recognition system that would recognize a common molecular pattern such as LPS found on gram negative bacteria. The part of the immune system that has this capability is called the innate immune system. The cells of the innate system (dendritic cells, macrophages, eosinophils, etc) have receptors (Toll-like Receptors 1-10 or TLRs) that recognize the common pathogen associated molecular patterns - PAMPs (sometimes called MAMP - microbe associated patterns), which leads to binding, engulfment, signal transduction, maturation (differentiation), antigen presentation, and cytokine/chemokine release from these cells. Take for example dendritic cells, which reside in the peripheral tissues and act as sentinels. They can bind PAMPs which include:
- CHO/Lipids on bacteria surface (LPS)
- mannose (CHO found in abundance on bacteria, yeast
- dsRNA (from viruses)
- nonmethylated CpG motiffs in bacterial DNA
TLR receptors are expressed on the cell surface for recognition of external PAMPS on foreign invaders. However, since bacterial and viral can be engulfed, it would be optimal to have intracellular recognition of viral and bacterial nucleic acids as well. These are recognized by intracellular TLRs in the cell after the they been taken up into the cells by endocytosis. The figure below shows how viral and bacterial nucleic acids found in endosomal vesicles, can be bound by endosomal membrane TLRs. A Jmol model of a recent structure of TLR3 and dsRNA is shown below.
Figure: Endosomal TLR3 Interaction with foreign RNA and DNA
![]() Jmol: Updated
TLR3:dsRNA complex
Jmol14 (Java) |
JSMol (HTML5) Needs updating
Jmol: Updated
TLR3:dsRNA complex
Jmol14 (Java) |
JSMol (HTML5) Needs updating
Inflammation can also arise when normal tissue is damaged due to injury, which exposes molecules usually located inside of the cell to the immune system. Such molecules include high mobility group proteins (associated with chromatin), proteoglycans and nucleic acids. These are referred to as damaged associated molecular pattern (DAMP) molecules. Intracellular proteins exists normally in a reducing environment so when they are exposed to the oxidizing conditions of the extracellular milieu, covalent and conformational changes may ensue that
Figure: Recognition of PAMPs by TLRs
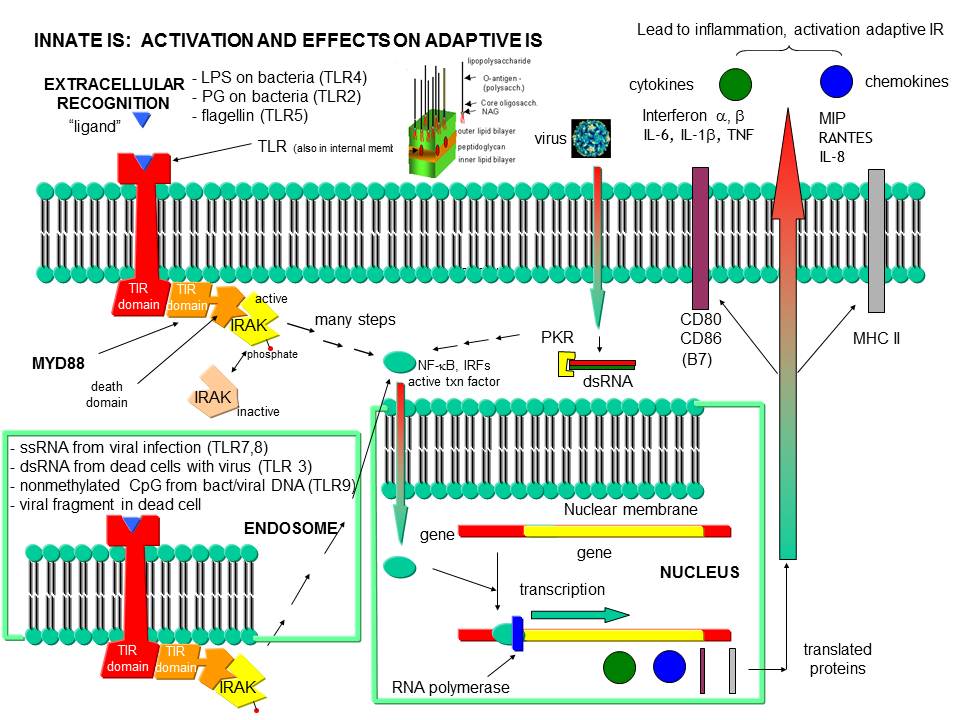{kind=link}

The figure below shows an overview of how the genetic information is decoded into proteins and how those processes are controlled.
An Overview of the Regulation of Gene Expression in Eukaroytes
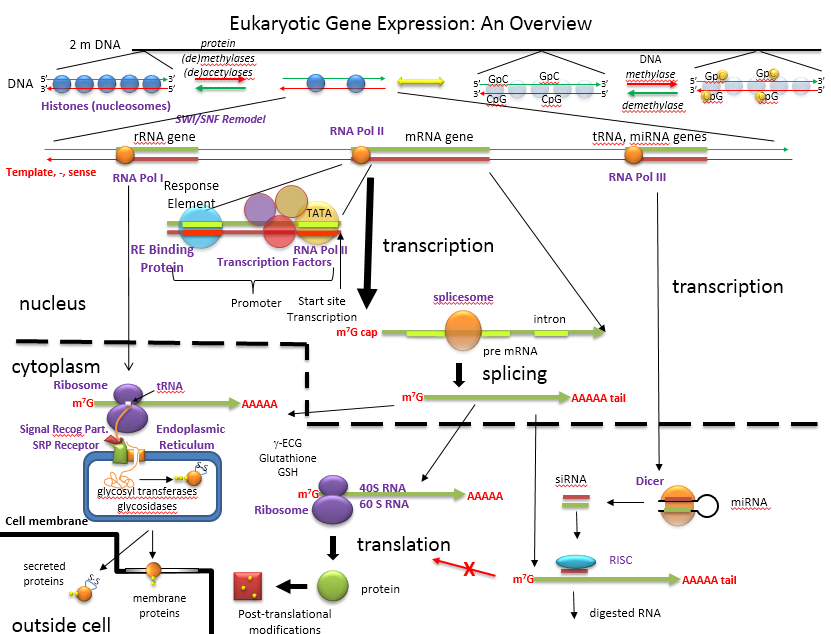
D15. ENCODE: Encyclopedia of DNA Elements
ENCODE is a public research consortium, sponsored by the National Human Genome Research Institute (NHGRI), who goals is to identify all functional elements in the human genome sequence. In September 2012, scientists involved in the project simultaneously published multiple papers the have significantly altered our understanding of how information about gene regulation is encoded into the genome. Scientists have now identified how function is encoded into most of the genome, a far cry from the early notion that nonprotein-coding DNA sequences, which comprise 98% of the genome, is irrelevant or junk DNA. Investigators used a variety of techniques to link structure to specific regions of the genome including sequencing of transcribed RNA from different types of cells, assessing senstivity/protection of DNA to an endonuclease, DNase 1, which gives information on DNA packing in chromatin and accessibility to transcription factors (see figure below), and mapping long range effects of distal enhancers on gene transcription,.
Figure: DNase 1 Sensitivity of Nucleosome Bound and Free DNA
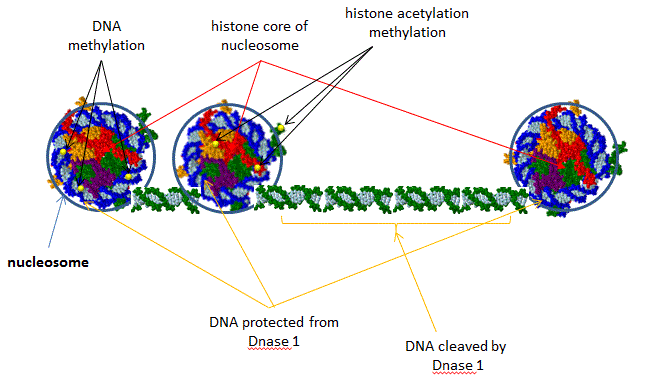
Here are some of the consortium's significant findings:
-
80% of the genome contains sequences linked to function;
-
The space between coding sequences contains a multitude of enhancer, promoters and sites of non-protein coding RNA transcription, many of which are in DNA regions shown to be involved in disease;
-
75% of DNA is transcribed during the life of the cell with transcription occurring from both strands and often overlapping;
-
Large numbers of DNase 1 sensitive sites map to experimental and theoretical sites for transcription factor binding;
-
A large number of sites correspond to motifs involved in DNA protein binding;
-
More than 1000 different distal sequences occur in any cell which affect gene transciption adding great complexity to the simple notion that binding of proteins at the promoter and proximal response elements controls gene transcription;
-
Cells have more than 200,000 DNase 1 sensitive site (far more than the number of promoter sites)
-
About 400,000 regions display "enhancer-like" properties and 70,000 display "promoter-like" properties;
-
Although transcription factors usually bind to under-methylated DNA, transcription factor binding was also found to inhibit methylation;
-
Regulation of transcription involves both binding of protein as well as RNA transcribed from non protein-coding regions of the DNA.
-
Evolutionary mutations in DNA regulatory sequences have potential evolutionary advantage over those in protein-coding sequences since regulatory effects on gene transcription are cell and time dependence, allowing changes in only certain cells at certain times compared to changes in a coding sequence which could affect many different cells times at many times.
-
Different RNA transcripts vary one million fold in their expression level.
This list clearly shows that genetic information encoded by the linear sequence of DNA ("1 bit" per 3.2 billion base pairs) is only a first approximation of the available information which is encoded by small contiguous stretches of DNA (as found in promoters), by loops distal to promoters (enhancers), chemical modification of DNA (methylation) and DNA binding proteins (methylation, acetylation, phosphorylation), and accessibility of DNA sequences to transcriptional regulators (RNA, protein) and packaging proteins (histones). All of these must be considered as we try to decode the human genome.
Just as a contrast, we have already discussed how proteins also have additional information elements other than their primary sequence which has the information necessary for protein folding. Short linear stretches of amino acids also act as signaling elements. These included N-terminal signal sequences which help locate proteins in the outside of the cell, degradation sequences (regions enriched in PEST amino acids - single letter code), signals that have evolved to encourage or discourage homodimer, heterodimer or nonspecific aggregation, as well as sequences that lead to specific post-translational modifications. Biological macromolecules (proteins and nucleic acids) are surely information macromolecules.
D16. Eukaryotic Species Complexity
The increasing complexity of eukaryotic organisms was thought to arise from an increasing number of genes. This simplistic assumptions has not been validated from the results of sequencing and annotating the genomes of many eukaryotic organisms. Compare these statistics: the number of putative genes in the simple nematode round worm C. Elegans, the fruit fly drosophila, and the human are approximately 20,000, 14,000, and about 23,000. There seems to be little correlation of species complexity with number of genes. Other possible mechanisms for increasing complexity from a given genome size include producing different proteins from the same genes through differential splicing of RNA transcripts and rearranging DNA as occurs in immune cells to produce the huge repertoire of possible antibody molecules necessary for recognition of nonself molecules (such as viruses and bacteria). These mechanisms can not account for the incredible complexity of the human species. Levine and Tjian have proposed two other mechanisms that could account for increasing complexity. Complexity would arise from the number of gene expression patterns and involve the involvement of nonprotein-coding regions of the genome, which in humans accounts for up to 98% of the genome. One mechanism requires the present of greater number and complexity of DNA regulatory sequences (enhancers, silencers, promoters) in more complex organisms. Since these sequences are in the DNA (the molecule that is transcribed), they are called cis-regulatory sequences. The second mechanism involves an increase in the elaboration and complexity of proteins (trans-regulatory elements) that regulate gene expression in more complex organisms.. These proteins could include transcription factors, proteins interacting with enhancer sequences, and proteins involved in chromatin remodeling (described above). They estimate that up to a third of the human genome (1 billion base pairs) might be involved in the regulation of gene transcription. In addition, 5-10% of all proteins expressed from genes appear to regulate gene transcription. There appears to be about 300, 1000, and 3000 transcription factor in yeast, drosophila and C. elegans, and humans, respectively. There is about one transcription factor for every gene in yeast, but one for every ten in humans.
In simple eukaryotes, cis regulatory elements would include the promoter (TATA box region), and upstream regulatory sequences (enhancer) and silencers about 100-200 base pairs from the promoter. In more complex eukaryotic species like humans, the promoter is more complex, containing the TATA box, initiator sequences (INR) and downstream promoter elements (DPE). Upstream cis regulatory elements (as far as 10 kb from the promoter) include multiple enhancers, silencers, and insulators. Most promoters have TATA boxes, where TATA Binding Protein (TBP) binds. Upstreams elements in turn regulate the binding of TBP.
- Eukaryotic promoters and regulatory regions
- Eukaryotic multisubunit general transcription apparatus
-
Biological
Regulation: BioBase - Gene Regulation (TRANS-FAC 6.0 public site
free with registration)
D17. Gene Expression Differences Between Humans and Chimpanzees
Our closest biological relative is the chimpanzee, who branched off from a common ancestor of both of us about six million years ago. Our DNA sequence appears to be 98.6 % identical (not just homologous). If we are so close in our genetic blue print, how can we be so different? There are many possible conjectures that can be answered by comparing the chimp and human genomes. Our genes are presumably very similar. People suspect that there are two major kinds of differences that make our species different:
- our genes are very similar but are transcribed differently in the two species. Recent evidence show that the types of RNA transcribed by human and chimp livers are very similar, but many more genes are transcribed in human brains compared to chimp brains.
- humans may have lost genes (or their function) that are required for chimp survival in the jungle. The observations that chimps are resistant to many of the disease pathogens that affect humans (immunodeficiency viruses like HIV, influenza A virus, hepatitis B/C, malarial parasite) could be explained by the loss of "protective" genes in humans. In addition, cardiovascular disease and certain types of cancer are rarer in chimps. Humans have apparently "lost" genes involved in body hair, strength, and early maturation, traits that would adapt the chimp to life in the jungle.
We previously discussed an example of a loss of gene function in humans. We have lost a hydroxylase gene involved in formation of certain types of sialic acids, specifically N-glycolylneuraminic acid, found on cell surface glycogroteins of mammals other than humans. Chimps have a lectin receptor for this sialic acid. Recent work has shown that human lack a critical Arg in our version of the lectin that would recognize N-glycolylneuraminic acid, making it unable to bind this ligand. Hence both pairs of genes involved in these type of interactions (cell:cell) are missing. Since sialic acid molecules are often involved in pathogen:host binding, these difference in humans compared to chimps might account for the difference in disease susceptibility as mentioned above.
{kind=link}
With respect to gene transcription in the brain, Lai et al. have found a mutation in the human gene FOXP2, a transcription factor, in a family that has significant difficulty in controlling muscles required for articulation of words. This mutation also causes problems in language processing and grammar construction. Comparison of the normal human gene with other primate genes shows distinct differences in the human gene which may have conferred on human the ability to use speech.
Chimp chromosome 22 is homologous to human chromosome 21. Recently, sequencers have found 1.44% single nucleotide changes between the two, a finding in line with overall homology between chimp and human DNA of 98.6%. The surprising finding was 68,000 insertions and deletions (indels) compare to humans. Most were short (<30 nucleotides). Those longer than 300 involved mobile genetic elements (transposons). Humans have a much higher incidence of insertions called Alu repeats. A high figure of 20% of homologous genes displayed significantly different expression levels.
In September 2005, a draft sequence of the chimpanzee genome and a comparison with the human genome was published by The Chimpanzee Sequencing and Analysis Consortium. Here are some of their findings:
- "single nucleotide substitutions occur at a mean rate of 1.23% between copies of the human and chimpanzee genome."
- "insertion and deletion (indel) events are fewer in number than single-nucleotide substitutions, but result in 1.5% of the euchromatic sequence in each species being lineage-specific."
- "There are notable differences in the rate of transposable element insertions: short interspersed elements (SINEs) have been threefold more active in humans, whereas chimpanzees have acquired two new families of retroviral elements."
- "Orthologous proteins in human and chimpanzee are extremely similar, with ~29% being identical and the typical orthologue differing by only two amino acids."
Since their genomes are over 3 billion base pairs, a 2% difference would mean around 60 million differences. The actual number appears to be 35 million single nucleotide differences (not counting insertions and deletions). Most of these would be expected not to be in genes and have little overall effect on phenotypic differences between the species. Finding the critical difference will be time consuming, and may requiring the sequencing of other primate genomes.
In their summary of the finding, Li and Saunders discuss changes in nucleotides that are synonymous (no changes in amino acids in the protein) and nonsynonymous. If a region of a gene can not tolerate changes that lead to amino acid alterations (i.e the nucleotides are under significant selective pressure not to change), the nonsyonymous rate of substitution would be lower than the rate of synonymous change. If change can occur without structure/function loss in the protein, the two rates would be similar. Comparing over 13,000 gene pairs from both organisms, they found the nonsynonymous rate to be about 25% of the synonymous rate. Hence most of the genes are conserved between species and would not be expected to contribute to the phenotypic difference in the organisms. Of the genes that showed higher nonsynonymous rates, none were obviously linked to brain function, but many were involved in immune function.
The biggest differences between the genomes were insertions/deletions (indels, numbering around 5 million) and gene duplications, not single nucleotide mutation. Insertions are often of two classes. Insertions include duplication of DNA stretches and addition of transposons ("jumping" gene or moveable DNA elements). These can be small (such as Alu repeats) or long (such as L1 insertions). In the human genome there are 7000 Alu sequences but in chimps there are 2300. Both have about equivalent numbers of Li insertions. Given that we have lost some traits (such as hair and strength), perhaps some chimp genes were lost in the human genome by the presence of indels. 53 such genes human genes were found. Perhaps the biggest change between chimps and humans is altered gene expression, which was not studied in this paper.
In another study by Xiaoxia Wang et al, a comparison was made of "pseuodgene" in humans (genes that acquired mutations in the past that disrupted their expression as functional protein and compared them to corresponding genes in chimps that still maintain function (i.e. they lead to functional proteins). Analysis showed that the identified pseudogenes were not randomly distributed among different classes of genes. Rather, there were concentrated in genes encoding olfactory receptor proteins, bitter tastant receptors, and immune system genes. Homo sapiens have a much diminished sense of smell. Bitter receptors probably became less important as humans switched from plants which contain many bitter toxins to meats. They attribute changes in immune system genes to changes in environment which might lead to gene lose if the intensity of the immune response, and the balance of immune self and nonself recognition might be altered in different environmental conditions.
Another major difference has been noted in gene copy number. Work by Hahn et al shows that gene copy number between human and chimps differ by 6.4%. After diverging from a common ancestor, humans gained 689 copies of some genes, compared to 26 for chimps. Likewise, humans lost 86 copies of some genes compare to a lose in chimps of 729 copies.
What maintains species? Barriers to interspecies hybrids.
New species seem to arise, according to evolutionary theory, when members of a species become geographically isolated. Each separated population accrues different mutations in their genomes, which confer adaptive advantages to each population in their different environment. With a long enough divergence time, genetic barriers to the production of viable hybrids between the population develop, leading to the divergence of the populations into separate species. This rational explanation doesn't give a specific molecular mechanism causing hybrid failure. In the 1930s, Dobzhansky and Muller proposed that changes in two genes that produce proteins that interact could account for interspecies hybrid failure. These genes would presumably mutate at a faster rate than usual. Within a species, the two genes would co-mutate at similar rates to produce proteins that still interact, but fast evolutionary change in the others "soon to be new species" gene pair would make hybrids produced from mating infertile at best, or lethal.
Brideau et al. have found a gene pair, lethal hybrid rescue (Lhr), that in Drosophila simulans diverged functionally, and hybrid male rescue (Hmr) in Drosophila melanogaster, which also has diverged functionally. F1 hybrid male offspring from crosses died. The Hmr gene in D. melanogaster is a transcription factor. The hmr gene is one of the most rapidly evolving genes in the genome. The exact function of the Lhr gene is uncertain but is associated with condensed chromatin (heterochromatin).
D18. Links and References
- ENCODE explained. Nature 389, 52-55 (2012)
- The Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57-74 (2012)
- Meijsing, S. et al. DNA Binding Site Sequence Directs Glucorticoid Receptor Structure and Activity. Science 324, 407 (2009)
- Choudhary, C. et al. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 325, 834 (2009)
-
- Rhos, R. et al. The role of DNA shape in Protein-DNA recognition. Nature 461, 1248 (2009)
- Geurts, A. M. et. al. Knockout Rats via Embryo Microinjection of Zinc-Finger Nucleases. Science. 325, p 433 (2009)
- McGowan, P. et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience 12, 342 (2009)
- Rubartelli, A & Lotze, M. Inside, outside, upside down: damage-associated molecular pattern molecules (DAMPs) and redox. Trends in Immunology. 28, 431 (2007) doi:10.1016/j.it.2007.08.004
- Jensen, L. and Bork, P. Not Comparable, but Complementary. Science 322, 56 (2008)
- Yu, H et al. High-Quality Binary Protein Interaction Map of the Yeast Interactome Network. Science 322, 104 (2008)
-
- Tarassov, K. et al. Science 320, 1465 (2008)
- Couzin, J. Erasing MicroRNAs Reveals Their Powerful Punch. Science, 316, 530 (2007).
- Jeffery P. Demuth, Tijl De Bie, Jason E. Stajich, Nello Cristianini, Matthew W. Hahn. The Evolution of Mammalian Gene Families
Brideau, N. et al . Two Dobzhansky-Muller Genes Interact to Cause Hybrid Lethality in Drosophila. 314,1292 (2006)- Segal, E. et al. A genomic code for nucleosome positioning. Nature. 442, 772 (2006)
- Xiaoxia Wang et al. PLoS Biology, issue 3 (2006).
- The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 437, 69 (2005); Li W. H. & Saunders, M. The Chimpanzee and Us. Nature 437, 50 (2005).
- Garza, M. and Hampton, R. Sterol Sensor comes up for air. Nature 435, 37 (2005)
- Rando, O et al. Genome-Scale Identification of Nucleosome Positions in S. cerevisiae. Science 309, 626-630, (2005)
- Urnov, F.D. et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646 (2005)
- The International Chimpanzee Chromosome 22 Consortium. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature 429, 382 (2004)
- Levine, M. & Tjian, R. Transcription regulation and animal diversity. Nature. 424, pg 147 (2003)
- Kamath. R. et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 421, pg 231 (2003).
- Winkler. W et al. Thiamine derivative bind messenger RNAs directly to regulate bacterial gene expression. Nature, 419, pg 852, 890 (2002)
- Enard et al. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418, pg 869 (2002)
- Angata et al. A second uniquely human mutation affecting sialic acid biology. J. Biol. Chem. 276, pg 40282 (2001)
- Dobrosotskaya et al. Regulation of SREBP Processing and Membrane Lipid Production by Phospholipids in Drosophila. Science. 296, pg 879 (2002)
- Nohturfft, A. and Losick, R. Fats, Flies, and Palmitate. Science. 296, pg 857 (2002)
- Plasterk, R. RNA Silencing: The Genome's Immune System. Science. 296, pg 1263 (2002)
- Dennis, C. The Brave New World of RNA. Nature. 418, pg 122 (2002)
- Hannon, G. RNA Interference. Nature. 418, pg 245 (2002)
- Munshi et al. Enhancers and Gene Transcription. 293, pg 1054, 1133 (2001)
- Yanofsky et al. Turning Gene Regulation on its Head. (About W Operon). Science. 293 pg 2018 (2001)
- Gene Expression in human and chimp brains. Science. 292, pg 44 (2001)
- Reprograming X Inactivation (one of two X chromosomes in female cell must be silenced) Science. 290 pg 1518 (2000)
- Wei et al. One Man's Food (about regulation of cyto P450) expression. Nature. 407 pg 852, 920 (2000)
- Pham and SauerNew Insight into an old modification (Tx. factor TFIID chemically modifies a histone) Science. 289. pg 2290, 2357 (2000)
- Lemon et al. Specifying Transcription. Nature. 414. pg 858, 924 (2001)
- Nakayama et al. The Histone modification circus. (how Histone modification regulates gene silencing) Science. 292, pg 64 (2001)
- Croix et al. Genes Expressed in Human Tumor Endothelium. Science. 289, pg 1197 (2000)
- de Urquiza et al. Docosahexaenoic Acid, a ligand for the retinoid X receptor in mouse brain. (ligand-activated txn factor through orphan receptor). Science. 290. pg 2140 (2000)
- Jaime F. Mart�nez-Garc�a, Enamul Huq, and Peter H. Quail. Direct Targeting of Light Signals to a Promoter Element-Bound Transcription Factor. Science 288, pg 859 (2001)
- Nagatani, A. Lighting Up the Nucleus. Science, 288, pg 821, 2000)
- A silence that speaks volumes. (gene silencing by RNA interference) Nature, 404, pg 804 (2000)
- Xho et al. How do X chromosomes set boundaries. (Females have 2 X chromosomes, males one. Do keep gene dosage comparable, one X chromosome must be inactivated in female clles. How is that done? They found a protein - trans-acting factor - that key). Science. 295. pg 287, 345 (2002)
- Jacobs and Khorasanizadeh. Structure of HP1 Chromodomain Bound to a Lysine 9-methylated Histone H3 tail. Science. 295. pg 2080 (2002)
 Navigation
Navigation
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 5D: Binding and the Control of Gene Transcription

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.