Biochemistry Online: An Approach Based on Chemical Logic

CHAPTER 2 - PROTEIN STRUCTURE
C: UNDERSTANDING PROTEIN CONFORMATION
BIOCHEMISTRY - DR. JAKUBOWSKI
3/4/16
|
Learning Goals/Objectives for Chapter 2C: After class and this reading, students will be able to
|
In contrast to micelles and bilayers, which are composed of aggregates of single and double chain amphiphiles, proteins are covalent polymers of 20 different amino acids, which fold, to a first approximation, in a thermodynamically spontaneous process into a single unique conformation, theoretically at a global energy minimum. This chapter section will investigate the possible conformations available to proteins, just as we studied the conformations of free fatty acids and acyl chains in lipid aggregates. The next chapter section will discuss the actual processes of folding and of unfolding (denaturation), both in vitro and in vivo. Then we will discuss the thermodynamics and intermolecular forces which stabilize the folded (or native) shape and the unfolded (or denatured state) of proteins, in a fashion similar to how we discussed micelle and bilayer stability.
C1. Main Chain Conformations
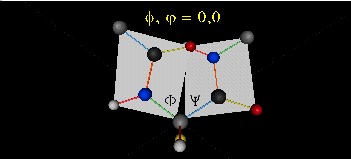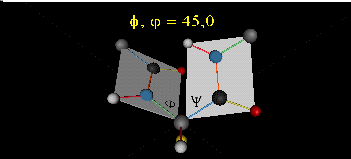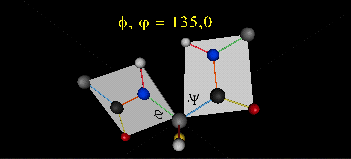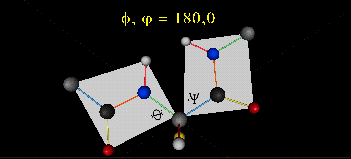
Just as saturated fatty acid chains have preferred conformations (all ttt), peptide chains also have preferred conformations. The complexity is much greater, however. With fatty acid chains, we dealt only with torsion or dihedral angles around the methylene carbons. For proteins, we must consider the covalent links which attach the amino acids together, as well as the rotations possible in 20 different amino acids. The peptide bonds connect the carbonyl C of the i th amino acid to the alpha amine N of the i th+1 amino acid. The resulting bond is an amide link. X-ray analysis shows that the the C-N bond has double bond character. This can be accounted for by delocalizing the nonbonding electron pair of the N to the carbonyl C forming a double bond, with the pi bonded electrons of the carbonyl C-O bond moving to the O. These resonance structures lead to a planar arrangement of the peptide carbonyl C and amide N and the two other atoms connected to each, since the hybridization of the C and N has sp2 character, with 120o bond angles. This greatly simplifies the number of conformations which a protein can adopt since these 6 atoms can be considered to reside and move in a plane. The alpha C serves as the corner attachment point of two different planes, each which can rotate independently of the other plane. The two planes can twist around the alpha carbon. The rotation angles for the two planes are called phi (f) and psi(y) are analogous to the torsion angles in the acyl chains of fatty acid. They can vary from -180 to +180o. The R group substituent attached to the alpha C can also rotate around the alpha C and the beta C of the side chain. This angle is defined as chi. Other rotations also occur within the side chain. We will concentrate on phi (f) and psi(y) angles in this section.
Figure: Extended Polypeptide Showing Planes and phi/psi Angles
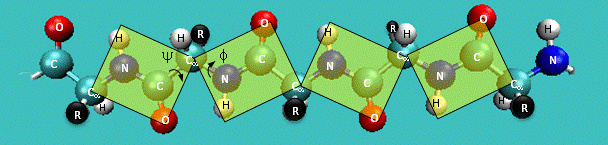
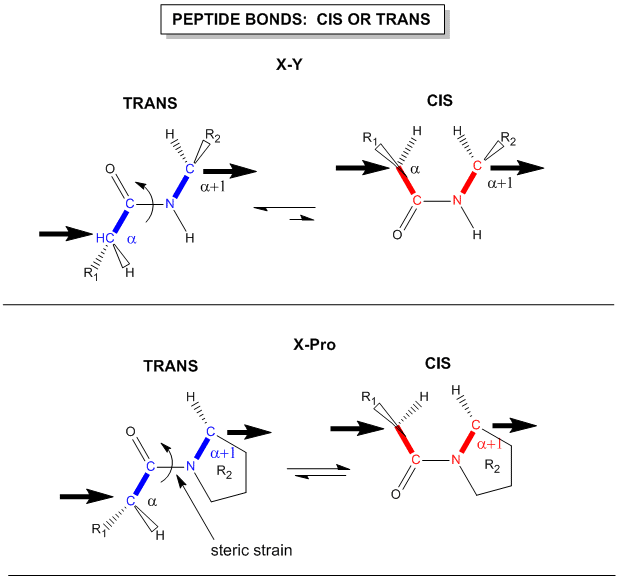
Another important feature of the peptide bond is that the alpha Cs at opposite ends of the rectangle are usually trans to each other (on opposite sides of the C-N bond in the peptide bond. This trans arrangement of the alpha Cs is sterically favored by a factor of 1000/1 for all peptide bonds except X-Pro. Pro, which is a cyclic amino acid, is sterically restricted.
Figure: trans arrangement of the alpha Cs
{kind=link}
![]()
The figure above, which also shows the X-Pro bond, clearly shows that both the trans and cis forms of the X-Pro bonds are hindered to a similar extent. In X-Pro bonds in proteins, the trans/cis ratio found in proteins is 4/1. The diagram above shows rans peptide bonds, and how they could be converted to cis through rotation around the C-N bond.
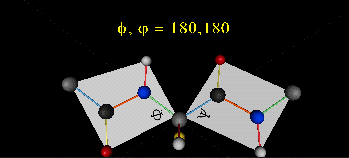
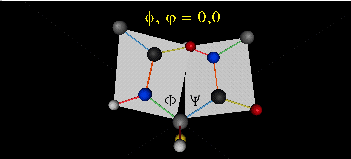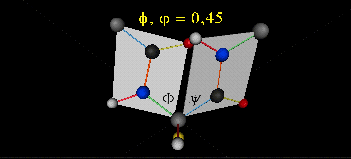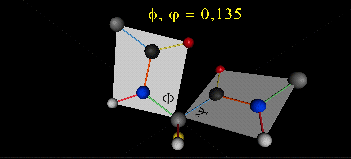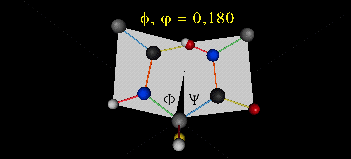
A protein can now be thought of as a series of linked sequences of rigid, planar peptide units which can rotate around phi/psi angles. When the chain is fully extended (as shown in the links above), phi/psi are 180o.
When phi (f) and psi(y) equal 0o, the two peptide bonds flanking the alpha Cs are in the same plane. This conformation is prohibited since the O of the C=O on one plane and the H of the H-N on the other are overlapping - i.e. they approach closer than their van der Waals radii.
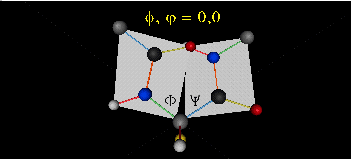
This simple example shows that all conformational space is not accessible for protein folding.
- Animation showing phi (f) angle rotation at psi (y)= 0.
- Animations showing psi (y) angle rotation at phi (f) = 0.
- Quick time movie (Alpha carbons are red. Move slider to approx. half way point to see "extended chain, analogous to the zig-zag conformation of the acyl chains in saturated fatty acids)
{kind=link}
{kind=link}
Ramachandran was the first to calculate which phi/psi angles are allowed. He modeled the angles permitted to a tripeptide, assuming the atoms were hard spheres. The angles allowed depended in part on the limiting distance chosen for interatomic contacts. (i.e. the usual H -- H distance is 2.0 angstroms, and 3.0 for C--C bonds.) The plot below show the allowed regions in red. Only 3 small regions of conformational space are available. If you allow a closer approach by 0.1 angstrom, more conformation space is available, but only one new area is available (shown in yellow in the plot below).
Figure: Ramachandran plot
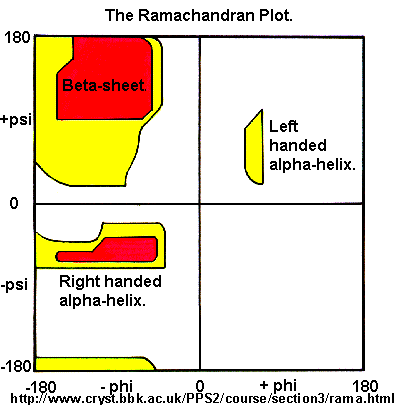
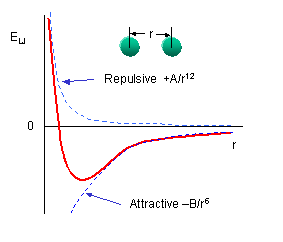
A Ramachandran plot of Ala-Ala-Ala is nearly identical to the plot for Phe-Phe-Phe (which is unbranched at the beta carbon (the first methylene C in the side chain). The plot for Thr-Thr-Thr, which has a branch at the beta C (with OH and CH3 attached) shows somewhat less room than the other plots. Pro-Pro-Pro is most restricted for obvious reasons. For a longer chain than a tripeptide, there are more restrictions than for (Ala)3, since the chain can't assume a conformation when it passes through itself. The plots for actual proteins have many points which do fall in forbidden regions. However, these points would be allowed if the peptide bonds is twisted a few degrees. Gly bonds also fall outside the allowed regions. This is understandable, since the side chain of Gly is H, and it is used in protein where sharp turns of the chain is necessary. Right hand alpha helices fall at -57,-47 while left hand alpha helices fall at +57,+47. (Notice these are not mirror images of each other. The mirror image of a right-handed alpha helix would be a left handed helix made of D-amino acids.) Parallel beta sheets are at -119, -113, while antiparallel sheets falls at -139, +135. Other types of helices also are found. The 310 helix , a sharper helix with 3 amino acids/turn, falls at -49,-26. All of these examples of secondary structure fall in allowed regions. Modern Ramachandran plots do not model the atoms as hard spheres but instead consider the potential energy of the atoms using the Lennard-Jones potential (6-12 potential) for van der Waals interactions. We discussed this potential function in the molecular modeling lab.
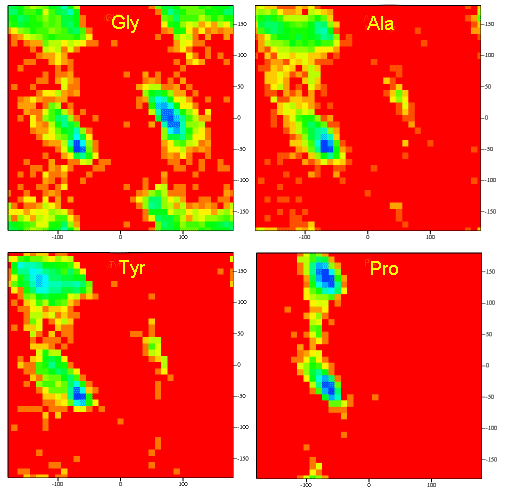
{kind=link}
Figure: Ramachandran plots showing phi/psi angles for Gly, Ala, Tyr, and Pro in actual proteins
-
 Ramachandran
Plots from Protopedia
Ramachandran
Plots from Protopedia -
Query
of Ramachandran Plots for any selected amino acid
-
Ramachandran
Plot Server
-
Secondary
Structure and Backbone Conformation from ExPASy.
(concentrate on first part on backbone conformation and Ramachandran
plots) (great site)
-
Side
Chain Conformations: How are side chain atoms and bond angles
designated? A tutorial from ExPASy
C2. Secondary Structure
Secondary structures are those repetitive structures involving H bond between amide H and carbonyl O in- the main chain. These include alpha helices, beta strands (sheets) and reverse turns..
Alpha Structure
Figure: Right Handed Alpha helices - image made with VMD
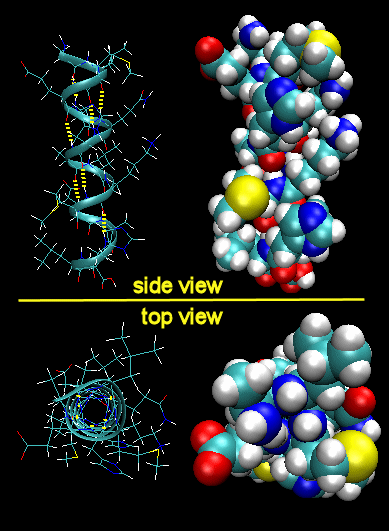
These helices are formed when the carbonyl O of the i th amino acid H bonds to the amide H of the i th +4 aa (4 amino acids away). The phi/psi angles for those amino acids in the alpha helix are - 57,-47, which emphasizes the regular repeating nature of the structure. It can also be characterized by n (the number of amino acid units/turn = 3.6) and pitch (the helix rise/turn = 5.4 angstroms). Some facts:
- the alpha helix is more compact than the fully extended polypeptide chain with phi/psi angles of 180o
- in proteins, the average number of amino acids in a helix is 11, which gives 3 turns.
- the left-handed alpha helix, although allowed from inspections of a Ramachandran plot, is never observed, since the side chains are too close to the backbone.
- the core of the helix is packed tightly. There are not holes or pores in the helix.
- All the R-groups extend backward and away from the helix axis.
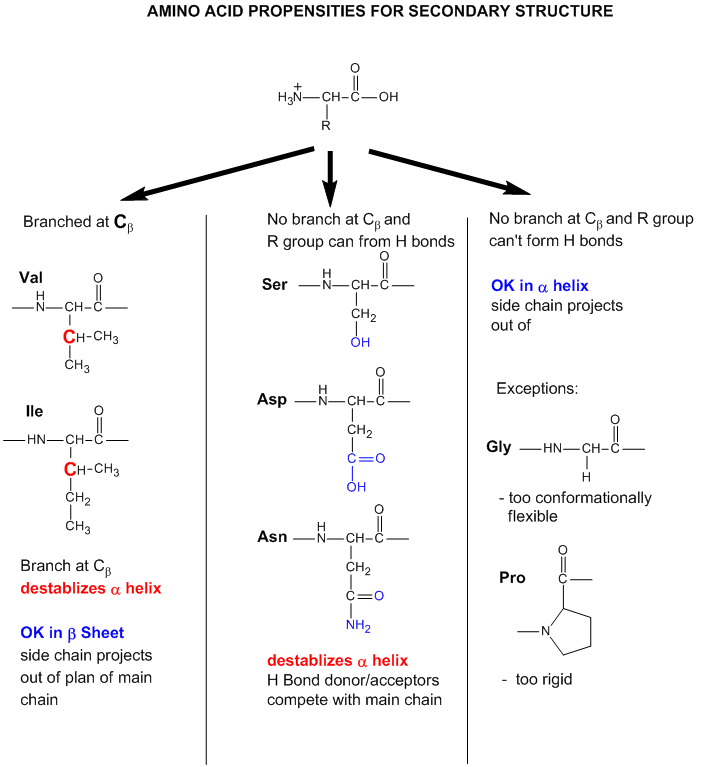
- Some amino acids are more commonly found in alpha helices than other. Amino acids can be divided into two kinds, those with branches at the beta C and those with none. Consider first those that aren't branched . Gly is too conformationally flexible to be found with high frequency in alpha helices, while Pro is too rigid. The amino acids with side chains that can H-bond (Ser, Asp, and Asn) and aren't too long appear to act as competitors of main chain H bond donor and acceptors, and destabilize alpha helices. The rest with no branches at the beta C can form helices. Those with branches at the beta carbon (Val, Ile) destabilize the alpha helix due to steric interactions of the bulky side chains with the helix backbone. (Remember left-handed alpha helices are not found in nature for similar reasons.) Summary of amino acids propensities for alpha helices (and beta structure as well)
- alpha keratins, the major component of hair, skin, fur, beaks, and fingernails, are almost all alpha helix.
![]() Jmol:
Updated An isolated helix from an Antifreeze Protein
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated An isolated helix from an Antifreeze Protein
Jmol14 (Java) |
JSMol (HTML5)
Note: There are other kinds of helices that can occur. These include a 310 helix and a p helix, which are stabilized by H-bonds between the amide NH and carbonyl O of residues (i, i+3) and (i, i+5), respectively. Likewise, they have 3 and 4.3 residues/turn, respectively, and a rise per residue of 6 and 4.7 angstrom, respectively. These structures are much rarer than right handed alpha helices.
| Helix Type | H bond btw ith and ith+X AA, where X = | Residue/turn | Rise (Angstrom)/turn |
| 310 | 3 | 3 | 6 |
| a | 4 | 3.6 | 5.4 |
| p | 5 | 4.3 | 4.7 |
Beta Structure
Beta Structure: Parallel and antiparallel beta strands are much more extended than alpha helices (phi/psi of -57,-47) but not as extended as a fully extended polypeptide chain (with phi/psi angles of +/- 180). The beta sheets are not quit so extended (parallel -119, +113 ; antiparallel, -139, +135), and can be envisioned as rippled sheets. They can be visualized by laying thin, pleated strips of paper side by side to make a "pleated sheet" of paper. Each strip of paper can be pictured as a single peptide strand in which the peptide backbone makes a zig-zag along the strip, with the alpha carbons lying at the folds of the pleats. Each single strand of the beta-sheet can be pictured as a twofold helix, i.e. a helix with 2 residues/turn. The arrangement of each successive peptide plane is pleated due to the tetrahedral nature of the alpha C. The H bonds are interstrand, not intrastrand as in the alpha helix.
Figure: Parallel beta strands (image made with Spartan)
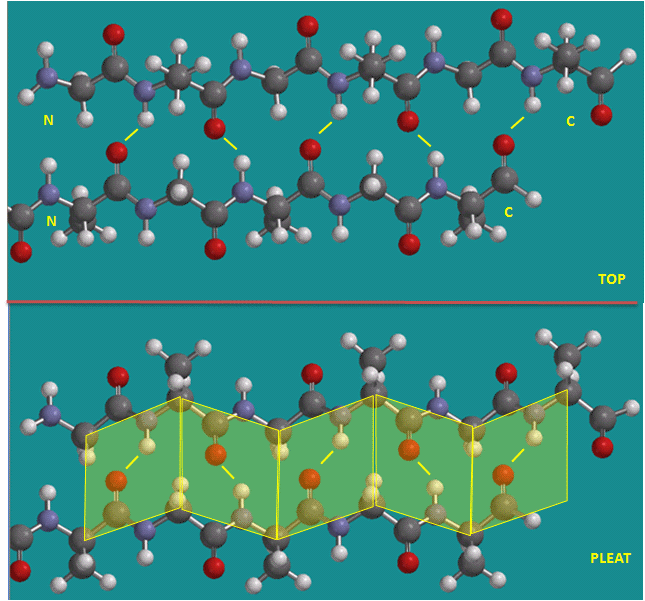
Figure: Antiparallel beta strands (image made with Spartan)

Note: Consider a strand as a continuous and contiguous polypeptide backbone propagating in one direction. Hence, using this definition, a helix consist of a single strand, and all the H-bonds are within the strand (or intrastrand). A beta sheet would then consist of multiple strands, since each "strand" is separated from other "strands" by an intervening contiguous stretch of amino acid which bends within the protein in a way which allows the next section of the peptide backbone, the next "strand", to H-bond with the first "strand". But remember, even in this case, all the H-bonds holding the alpha and beta structure together are intramolecular.
In a parallel beta sheet structure, the optimal H bond pattern leads to a less extended structure (phi/psi of -119, +113) than the optimal arrangement of the H bonds in the antiparallel structure (phi/psi of -139, +135). Also the H bonds in the parallel sheet are bent significantly. (i.e. the carbonyl O on one strand is not exactly opposite the amide H on the adjacent strand, as it is in the antiparallel sheet.) Hence antiparallel beta strands are presumably more stable, even though both are abundantly found in nature. Short parallel beta sheets of 4 strands or less are not common, which might reflect their lower stability.
The side chains in the beta sheet are normal to the plane of the sheet, extending out from the plane on alternating sides. Parallel sheets characteristically distribute hydrophobic side chains on both side of the sheet, while antiparallel sheets are usually arranged with all the hydrophobic residues on one side. This requires an alternation of hydrophilic and hydrophobic side chains in the primary sequence. Antiparallel sheets are found in silk with the sheets running parallel to the silk fibers. The following repeat is found in the primary sequence: (Ser-Gly-Ala-Gly)n), with Gly pointing out from one face, and Ser or Ala from the other.
![]()
![]() Jmol:
Silk
Jmol:
Silk
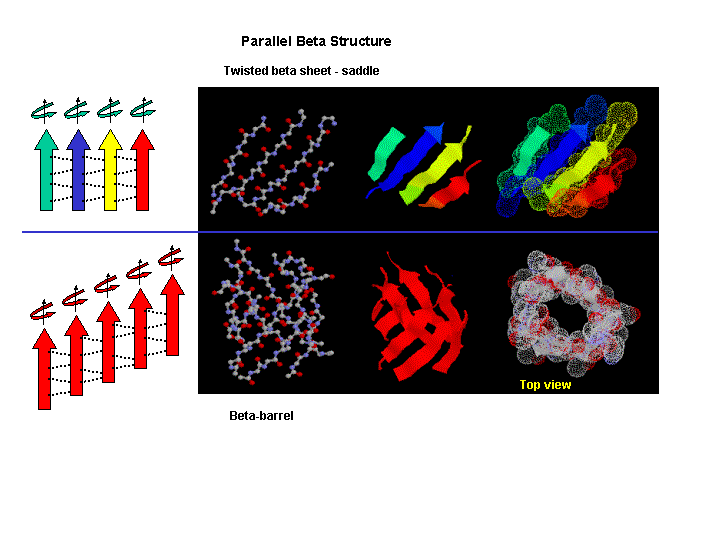
Beta strands have a tendency to twist in the right hand direction. This leads to important consequences in how the beta strands are connected. Parallel strands can form twisted sheets or saddles as well as beta barrels.
{kind=link}
Figure: Twisted Beta Sheet/Saddle (image made with VMD)
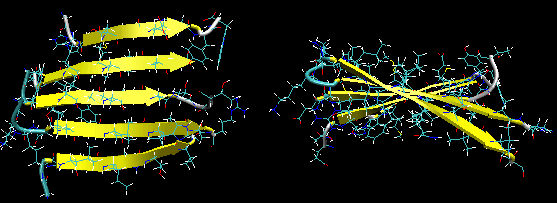
Figure: Beta Barrel (image made with VMD)

- in parallel strands, right handed connectivity is common.
- in a protein with parallel strand in register, and given the inherent twist in the stands, the strands arrange in a way to have the H bonds stretched equally at the ends of the chains, giving rise to a twisted saddle shape (top structure above).
![]() Jmol:
Updated Twisted beta sheet from Arabinose Binding Protein
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Twisted beta sheet from Arabinose Binding Protein
Jmol14 (Java) |
JSMol (HTML5)
- in a protein with parallel strand out of register, and given the inherent twist in the stands, the strands arrange in a way to have the H bonds stretched equally at the ends of the chains, giving rise to a beta barrel (bottom structure above).
![]() Jmol:
Beta barrel from triose phosphate isomerase
Jmol:
Beta barrel from triose phosphate isomerase
Reverse Turns: About 50% of the amino acids in a globular protein are in regular secondary structure (alpha or beta). The remaining amino acids are not less ordered, just less regular. An additional example of secondary structures is reverse turns (or beta-bends or beta turns). Reverse turns often connect successive antiparallel beta strands and are then called beta hairpins.
Figure: Reverse Turns (image made with VMD)
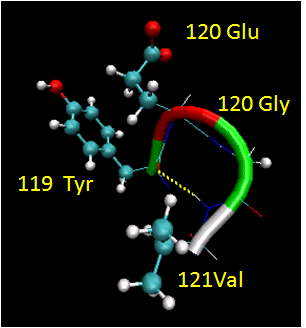
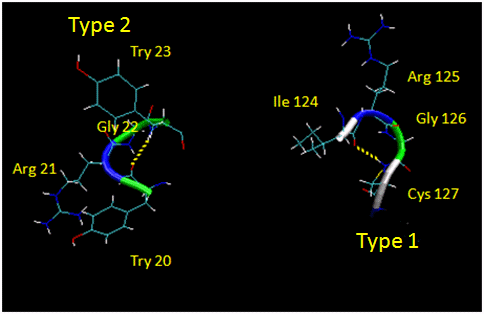
They are almost always at the surface, and consist of 4 amino acids. There are two types. (I - f2 = -60, y2=-30; f3 = -90, y3 = 0; II - f2 = -60, y2=120; f3 = 90, y3 = 0 ) Residue 2 of both is often Pro. (Why?) Both have an H bond between the carbonyl O of the i th a.a and the amide H of the i th+3 aa (three amino acids away). In the type 2, the O of residue 2 crowds the beta C of residue 3, so aa2 is usually Gly. Why? Those amino acids which destabilize alpha helices are often found in beta sheets, since the side chains project out of the plan which holds the main chain.
Figure: Type I and Type II Reverse Turns - from hen egg white lysozyme (image make with VMD)
![]() Jmol:
Updated Reverse Turn: Trypsin Inhibitor
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Reverse Turn: Trypsin Inhibitor
Jmol14 (Java) |
JSMol (HTML5)
(Notice the tightness of the reverse
turn and the presence of Pro and
Gly.)
Figure: Why do amino acid propensities for secondary structure differ?
-
Secondary
Structure and Backbone Conformation from
ExPASy.(concentrate on second part on secondary structure)
C3. Tertiary Structure
To a first approximation, you may think the a globular protein might fold such thal all the hydrophobic side chains are buried in the interior of the protein, surrounded by other hydrophobic side chains. In a similar fashion, you might expect the polar and charged side chains could be on the surface, exposed to water. Such a model would be analogous to a micelle which has an almost "perfect" separation of polar (on the surface) and nonpolar atoms (buried). If it were only that simple!. Topologically, it is impossible for a protein to fold in an intramolecular fashion in strict analogy to the the intermolecular aggregation of single chain amphiphiles into a micelle. Consider also that the entire backbone is polar. To a first approximation we would expect the bulk nonpolar groups would be buried surrounded by other nonpolar groups. Likewise we would expect the bulk of polar and charged would be on the surface. The Jmol models below show the similarities in the formations of a micelle, in which all nonpolars are buried, to that of protein in which most nonpolar side chain are buried and surrounded in a nonpolar environment.
![]() Jmol:
Updated A protein with a buried nonpolar amino acid
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated A protein with a buried nonpolar amino acid
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol:
Updated Micelle
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated Micelle
Jmol14 (Java) |
JSMol (HTML5)
What is the preferred disposition of side chains in proteins as evidenced from the crystal structure of thousands of proteins? Here are some conclusions from a paper by Pace (Biochemistry. 40, pg 310 (2001)
- On average, about 50% of the amino acids are in secondary structure. On average, there is about 27% alpha helix, and 23% beta structure. Of course, some proteins are almost all alpha-helical, and some are almost all beta structure, but most are a mixture.
- The side chain location varies with polarity. Nonpolar side chains, such as Val, Leu, Ile, Met, and Phe are predominately (83%) in the interior of the protein.
- Charged polar side chains are almost equally partitioned between being buried or exposed on the surface. (54% - Asp, Glu, His, Arg, Lys are buried away from water, a bit startling)
- Uncharged polar groups such as Asn, Gln, Ser, Thr, Tyr are mostly (63%) buried, and not on the surface (a bit startling) .
- Globular proteins are quite compact, with water excluded. The packing density (Vvdw/Vtot) is about 0.75, which is like the NaCl crystal and equals the closest packing density of 0.74. This compares to organic liquids, whose density is about 0.6-0.7.
Tertiary structure and pKas
If a charged size chain is buried in a protein, you would expect that it would be surrounded, in general, by either oppositely charged side chains, to which it could form an internal salt bridge (ion-ion interaction), or a polar uncharged group with which it could interact through dipole-dipole or, more specifically, H bond interactions. You would also expect that if it were not near an oppositely charged side chain, that it would exist, if buried, in an uncharged state.
Hence the pKa of side chains would be dramatically affected by the nature of its microenvironment (as we have already seen with the pKa of acetic acid in solvents of different polarity). NMR spectroscopy has been used to determine the pKa values of specific side chains in protein whose crystal structure is known. Pace et al (2009) summarize data on the properties of ionizable side chains in a series of proteins whose structure has been determined. The intrinsic pKa, pKaint or prototypical pKa value for a side chain exposed to water can be determined using a pentapeptide containing the target amino acid X surrounded by 2 Ala ion either each side with both the N and C termini of the peptide blocked so they are uncharged.
Table: pKa values of ionizable side chains in a series of protein
| Group | Content % | Buried % | pKaint in AAXAA | pKa avg | low pKa | high pKa | # measurments |
| Asp | 5.2 | 56 | 3.9 | 3.5 + 1.2 | 0.5 | 9.2 | 139 |
| Glu | 6.5 | 48 | 4.3 | 4.2 + 0.9 | 2.1 | 8.8 | 153 |
| His | 2.2 | 72 | 6.5 | 6.6 + 1.0 | 2.4 | 9.2 | 131 |
| Cys | 1.2 | 90 | 8.6 | 6.8 + 2.7 | 2.5 | 11.1 | 25 |
| Tyr | 3.2 | 67 | 9.8 | 10.3 + 1.2 | 6.1 | 12.1 | 20 |
| Lys | 5.9 | 34 | 10.4 | 10.5 + 1.1 | 5.7 | 12.1 | 35 |
| Arg | 5.1 | 56 | 12.3 | ||||
| C term | 3.7 | 3.3 + 0.8 | 2.4 | 5.9 | 22 | ||
| N term | 8.0 | 7.7 + 0.5 | 6.8 | 9.1 | 16 |
A quick glance a the table shows a huge variation in the pKas of ionizable side chains in proteins with the pKa of Asp varying over a range of 8.7 pH units, showing that it can act at physiological pH as either a strong acid or a moderate base. Three majors effects can perturb the pKa of ionizable side chains:
1. Dehydration of side chain as it is buried in a protein (Born Effect): The stability of a charged group depends on the polarity of the medium in which it exists. Ions are more stable in water than in nonpolar solvents as the water molecules can reorient and interact with the ion through ion-dipole or ion-H bond interactions, which effectively shields the ion from other counter ion. The shielding effect of water is related to the dielectric constant, e, of the solvent. Coulombs law can be written as Fcoul = q1q2/er2 which can be expressed in energy terms as DGcoul = q1q2/er. Epsilon is the dielectric constant of the solvent. Water has a higher dlelectric constant (80) than nonpolar solvents (4-10) and hence shields opposing charges more, stabilizing them. Hence the pKa of side chains of those amino whose deprotonated state is charged will have their pKa values raised (so they are less acidic) in nonpolar environments. The reverse holds for side chains whose protonated form is charged. Pace cites as an example two mutant of staphylococcal nuclease in which a buried Val 66 is changed either to Asp or Lys. The buried Asp has a pKa of 8.9 compared to 5.5 for the buried Lys. These changes were not compensated for with new charge-charge interactions, so the change can be attributed to the dehydration (or Born) effect.
2. Ion-Ion interactions with another charged side chain through Coulombic forces: This effect can be most readily observed at the surface of the protein. Pace cites a study of RNase Sa that is devoid of Lys and has a pI of 3.5. Five Asp and Glu were replaced on the surface using site-specific mutagenesis with Lys, which changed the pI of the protein to 10.2. At pH 7, the protein without Lys had a charge of -7 while the protein with 5 Lys had a charge of +3. The crystal structures were similar so Coulombic interactions would determine the differences in the pKa of the 11 common side chains. On average the mutant pKas were higher by 0.75 pH units, which makes sense as the mutant had a high pI. Calculated pKa values were similar to those determined by NMR. These data are consistent with the idea that Coulombic interactions are the chief cause of pKa changes in surface side chains.
3. Charge-dipole interactions and H bonds: It should be obvious that charge states of ionizable side chains would be adjusted to optimize H bond (and more generally charge-dipole) interactions in proteins. If the interactions are optimal in the charged state, pKa values for His and Lys would be increased and for Asp, Glu, Cys, and Tyr they would be decreased. Pace cites the buried Asp 76 in RNase T1 in which the Asp is charged but does not form an internal salt bridge. It has a depressed pKa of 0.6 and has 3 H bonds to the side chains of Asn 9, Tyr 11 and Thr 91. Mutants were made to remove the H bonds to see the effect on the pKa of Asp 76. Removing 1, 2, or 3 H bonds changed the pKa to 3.3, 5.1, and 6.4 respectively. The 6.4 value is much higher than the pKint, which can be attributed to the Born effect.
C4. Common Motifs in Proteins
Super-Secondary Structure - Given the number of possible combinations of 1o, 2o, and 3o structures, one might guess that the 3D structure of each protein is quite distinctive. This is true. However, it has been found that similar substructures are found in proteins. For instance, common secondary structures are often grouped together to form a motifs called super-secondary structure (SSS). See some examples below:
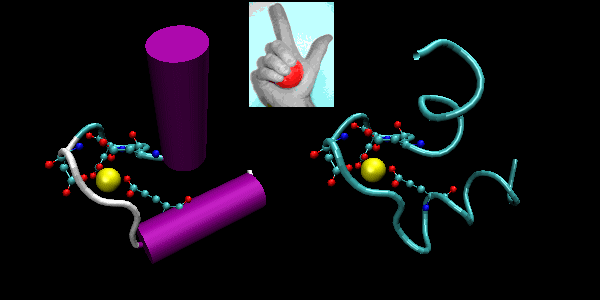
- helix-loop-helix : found in DNA binding proteins and also in calcium binding proteins. This motif, which is also a helix-loop-helix, is often called the EF hand. The loop region in calcium binding proteins are enriched in Asp, Glu, Ser, and Thr. Why? The EF hand shown below is from calmodulin.
Figure: helix-loop-helix (image made with VMD)
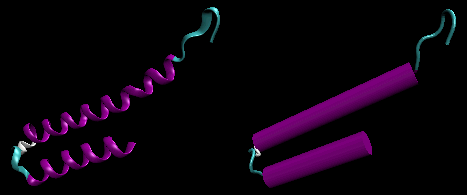
Figure: EF Hand
![]() Jmol:
Updated helix-loop-helix of the lambda Repressor
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated helix-loop-helix of the lambda Repressor
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol:
Updated helix-loop-helix (EF hand) from calmodulin
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated helix-loop-helix (EF hand) from calmodulin
Jmol14 (Java) |
JSMol (HTML5)
- beta-hairpin or beta-beta: is present in most antiparallel beta structures both as an isolated ribbon and as part of beta sheets.
Figure: beta-hairpin, or beta-beta (image made with VMD)

![]() Jmol:
Updated beta-hairpin from bovine pancreatic trypsin inhibitor
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated beta-hairpin from bovine pancreatic trypsin inhibitor
Jmol14 (Java) |
JSMol (HTML5)
- Greek Key motif: four adjacent antiparallel beta strands are often arranged in a pattern similar to the repeating unit of one of the ornamental patterns used in ancient Greece.
Figure: Greek Key Motiff
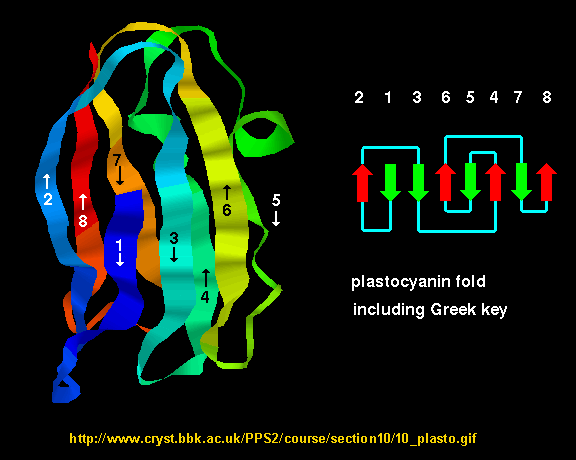
![]() Jmol:
Greek Key
Jmol:
Greek Key
- Figure: beta-alpha-beta: is a common way to connect two parallel beta strands. (beta hairpin used for antiparallel beta strands).
Figure: beta-alpha-beta (image made with VMD with H atoms added by Molprobity
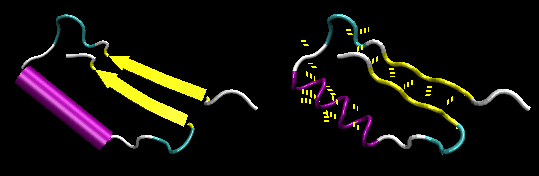
![]() Jmol: Updated
beta-helix-beta motif from triose phosphate isomerase
Jmol14 (Java) |
JSMol (HTML5)
Jmol: Updated
beta-helix-beta motif from triose phosphate isomerase
Jmol14 (Java) |
JSMol (HTML5)
-
Beta Helices: These right-handed parallel helix structures consists of a contiguous polypeptide chain with three parallel beta strands separated by three turns forming a single rung of a larger helical structure which in total might contain as many as nine rungs. The intrastrand H-bonds are between parallel beta strands in separate rungs. These seem to prevalent in pathogens (bacteria, viruses, toxins) proteins that facilitate binding of the pathogen to a host cell.
Figure: Beta Helices (image made with VMD)
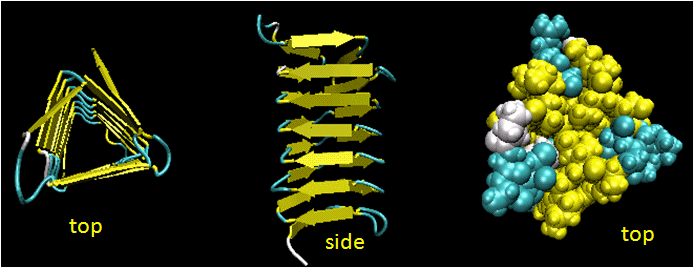
Table: Beta Helices
Vibrio cholerae
cholera
Helicobacter pylori
ulcers
Plasmodium falciparum
malaria
Chlamyidia trachomatis
VD
Chlamydophilia pneumoniae
respiratory infection
Trypanosoma brucei
sleeping sickness
Borrelia burgdorferi
Lymes disease
Bordetella parapertussis
whooping cough
Bacillus anthracis
anthrax
Neisseria meningitides
menigitis
Legionaella pneumophilia
Legionaire�s disease
-
of
the Swiss Institute of Bioinformatics.
(SIB) is dedicated to the analysis of protein sequences and structures
as well as 2-D PAGE
Domains -
Domains are the fundamental unit of 3o structure. It can be considered a chain or part of a chain that can independently fold into a stable tertiary structure. Domains are units of structure but can also be units of function. Some proteins can be cleaved at a single peptide bonds to form two fragments. Often, these can fold independently of each other, and sometimes each unit retains an activity that was present in the uncleaved protein. Sometimes binding sites on the proteins are found in the interface between the structural domains. Many proteins seem to share functional and structure domains, suggesting that the DNA of each shared domain might have arisen from duplication of a primordial gene with a particular structure and function.
Evolution has led towards increasing complexity which has required proteins of new structure and function. Increased and different functionalities in proteins have been obtained with additions of domains to base protein. Chothia (2003) has defined domain in an evolutionary and genetic sense as "an evolutionary unit whose coding sequence can be duplicated and/or undergo recombination". Proteins range from small with a single domain (typically from 100-250 amino acids) to large with many domains. From recent analyzes of genomes, new protein functionalities appear to arise from addition or exchange of other domains which, according to Chothia, result from
- "duplication of sequences that code for one or more domains
- divergence of duplicated sequences by mutations, deletions, and insertions that produce modified structures that may have useful new properties to be selected
- recombination of genes that result in novel arrangement of domains."
Structural analyses show that about half of all protein coding sequences in genomes are homologous to other known protein structures. There appears to be about 750 different families of domains (i.e small proteins derived from a common ancestor) in vertebrates, each with about 50 homologous structures. About 430 of these domain families are found in all the genomes that have been solved.
C5. Structual Clases of Proteins
Proteins can be divided into 3 classes of protein, depending on their characteristic secondary structure. Click below for Chime structures showing examples of these proteins.
- alpha proteins - consist of predominately alpha helix.
![]() Jmol:
Updated cytochrome B562
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated cytochrome B562
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol:
Updated met-myoglobin
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated met-myoglobin
Jmol14 (Java) |
JSMol (HTML5)
- alpha/beta proteins - consist of a common of alpha and beta structure. These are the most common class.
![]() Jmol:
Updated triose phosphate isomerase
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated triose phosphate isomerase
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol:
Updated hexokinase
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated hexokinase
Jmol14 (Java) |
JSMol (HTML5)
- beta proteins - consist of predominantly beta structure.
![]() Jmol:
Updated superoxide dismutase
Jmol14 (Java) |
JSMol (HTML5)
Jmol:
Updated superoxide dismutase
Jmol14 (Java) |
JSMol (HTML5)
![]() Jmol:
Updated human IgG1 antibodyJmol14
(Java) |
JSMol (HTML5)
Jmol:
Updated human IgG1 antibodyJmol14
(Java) |
JSMol (HTML5)
![]() Jmol:
Updated retinol binding proteinJmol14
(Java) |
JSMol (HTML5)
Jmol:
Updated retinol binding proteinJmol14
(Java) |
JSMol (HTML5)
fatty acid binding proteins; Peptide-N(4)-(N-Acetyl-b-D-Glucosaminyl) Asparagine Amidase (PNGase F) - under construction.
A more complete classification of protein structure has been developed based on the following hierarchy of organization: Class, Architecture, Topology, and Homologous Superfamilies - CATH.
- Class: the highest level of organization which consists of four classes - mainly alpha, mainly beta, alpha-beta, and few secondary structures
- Architecture (40 types): describes the shape of domain based on secondary structures but doesn't describe how they are connected. Ex: beta barrel, beta propellor
- Topology (or fold group, 1233 types): members in topology groups have a common fold or topology in the "core" of the domain structure.
- Homologous Superfamilies (2386 types): These groups are homologous in sequence or structure and derive from a common precursor gene/protein.
Structural Biology Knowledge Base
Here are some 3D structures resources, accessible through a sequence or ID-based search. and collated in Nature's Structural Biology Knowledge Base.
-
CATH - structural classification of manually curated classification of protein domain structures
-
DisProt - Database of Protein Disorder
-
Gene3D - CATH domain assignments for protein sequences
-
NESG Functional Annotation Database - Computational analysis of function of protein of unknown function
-
RCSB PDB - Protein Data Bank USA
-
PDBe - Protein Databank Europe
-
PDBj - Protein Databank Japan
-
PDBsum - a pictorial database that provides an at-a-glance overview of the contents of each 3D structure deposited in the Protein Data Bank
-
PROCOGNATE - database of cognate ligands for the domains of enzyme structures in CATH, SCOP and Pfam
-
SCOP - Structural Classification of Proteins: detailed and comprehensive description of the structural and evolutionary relationships between all proteins whose structures are known
-
SMART (Simple Modular Architecture Research Tool) - allows the identification and annotation of genetically mobile domains and the analysis of domain architectures
C6. Quarternary Structure
Primary structure is the linear sequence of the protein. Secondary structure is the repetitive structure formed from H-bonds among backbone amide H and carbonyl O atoms. Tertiary structure is the overall 3D structure of the protein. Quaternary structure is the overall structure that arises when tertiary structures aggregate to self to form homodimers, homotrimers, or homopolymers OR aggregate with different proteins to form heteropolymers.
-
Quaternary
Structure from ExPAYs
Globular versus fibril structures
We will deal exclusively with proteins which have a "globular" tertiary structure in this course. However, there are many proteins that form elongated fibrils with properties like elasticity, which measures the extent of deformation with a given force and subsequent return to the original state. Elastic molecules must store energy (go to a higher energy state) when the elongating force is applied, and the energy must be released on return to the equilibrium resting structure. Structures that can store energy and release it when subjected to a force have resiliency. Proteins that stretch with an applied forces include elastin (in blood vessels, lungs and skins where elasticity is required), resilin in insects (which stretches on wing beating), silk, found in spider web) and fibrillin found in most connective tissues and cartilage. Some proteins have high resiliency (90% in elastin and resilin), while others are only partially resilient (35% in silk, which have a tensile strength approaching that of stainless steel. In contrast to rubber, which has an amorphous structure which imparts elasticity, these proteins, although they have a dissimilar amino acid sequence, seem to have a common structure inferred from their DNA sequences. In some (like fibrillin), the protein has a folded b-sheet domain which unfold like a stretched accordion. Others (like elastin and spider silk) have b-sheet domain and other secondary structures (a-helices and (b turns) along with Pro and Ala repetitions. Researcher are studying these structures to help in the synthesis of new elastic and resilient products
-
SCOP:
Structural Characterization of Proteins - Database showing folds,
superfamiles, families, and domains
C7. Recent References
- Pace, C. et al. Protein Ionizable Groups: pK values and Their Contribution to Protein Stability and Solubility. J. Biol Chem. 284, 13285 (2009)
- Chothia, C. et al. Evolution of Protein Repertoire. Science, 300, pg1701 (2003)
- Stebbins & Galan. Structural Mimicry in Bacterial Virulence . Nature. 416. pg 701 (2001)
- Taylor. A deeply knotted protein structure and how it might fold. Nature. 406. pg 916 (2000)
- Innate immunity: ancient system gets new respect (about antimicrobial peptides). Science. 291 pg 2068 (2001)
- Graether et al. b-helix structure and ice-binding properties of a hyperactive antifreeze protein from an insect. Nature. 406, pg 249, 325 (2000); Liou et al. Mimicry of ice structure of surface hydroxyls and water of a b-helix antifreeze protein. Nature. 406, pg 322,(2000)
- Kanamaru. S et al. Structure of the cell-puncturing device of Bacteriophage T4. Nature. 415. pg 553 (2002)
 Navigation
Navigation
Return to Biochemistry Online Table of Contents
Archived version of full Chapter 2C: Understanding Protein Conformation

Biochemistry Online by Henry Jakubowski is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.